For the following sequence -
SGIVKMVSPTSKVEPCIVSVTYGNMTLNGLWLD
DKVYCPRHVICSSADMTDPDYPNLLCRVTSSDF
CVMSGRMSLTVMSYQMQGCQLVLTVTLQNPNTP
KYSFGVVKPGETFTVLAAYNGRPQGAFHVTLRS
SHTIKGSFLCGSCGSVGYVLTGDSVRFVYMHQL
ELSTGCHTGTDFSGNFYGPYRDAQVVQLPVQDY
TQTVNVVAWLYAAIFNRCNWFVQSDSCSLEEFN
VWAMTNGFSSIKADLVLDALASMTGVTVEQVLA
AIKRLHSGFQGKQILGSCVLEDELTPSDVYQQL
AGVKLQ
(a) Please identify this protein.
1. Goto NCBI Homepage, then select BLAST icon in the top area, then in the BLAST page, select Protein-Protein BLAST(blastp).
2. In the blastp page, enter the given sequence in the dialogue box like this,
then press "BLAST!" button. In the following page, press the "FORMAT!"
button as well.
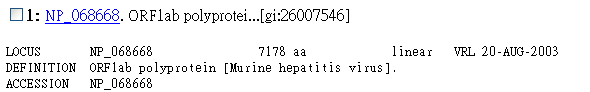
3. Then you should see some results coming out. We could see from the results
that both the first two sequences has E-values 0, which means exact match. But
if we further examine these two entries, we will find that entry 1 is only an
ORF entry, but entry 2 is a RNA-directed RNA polymerase, which is very likely
the protein we want.

(b) Can you find 3D structure for this protein?
1. No.
(c) If not, can you make a 3D structure model for this protein? Use three pictures to show your model.
1. Yes. Goto ExPASy homepage, and select SWISS-MODEL from Tools and software package.
2. In the Swiss model page, select the "First-Approach mode".
3. Enter the protein sequence in the sequence dialogue box, and set the parameters below and then submit.
4. Or you could select a template for homology modeling, select the SWISS-MODEL
BLAST link, enter the sequence in the dialogue box and then press submit
button. After results showing up, record the first ExPDB ID and input it in
the First-Approach mode page's template section.

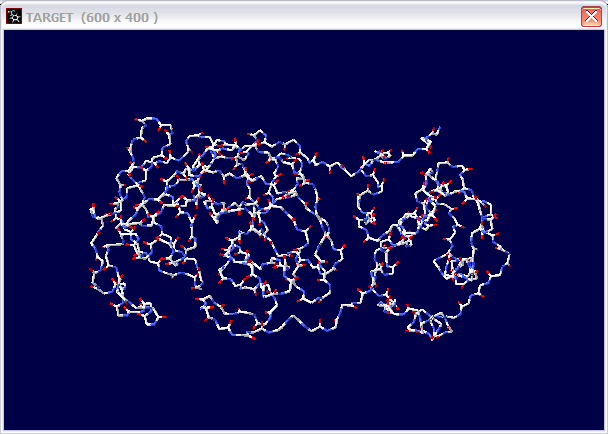
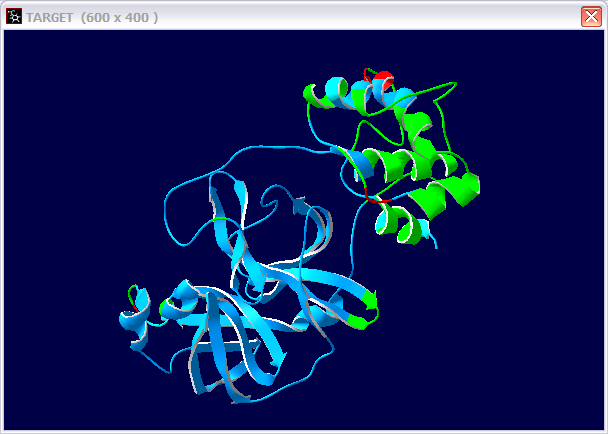
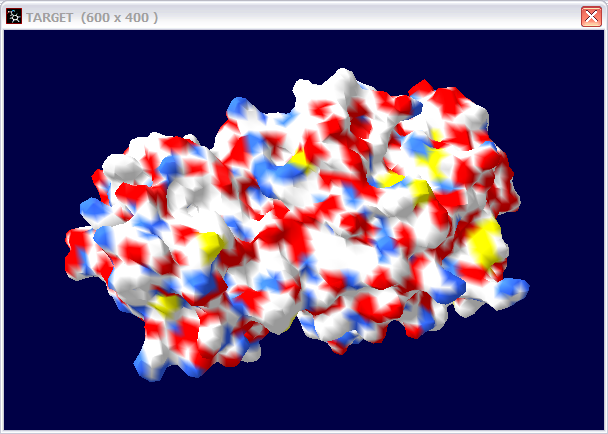
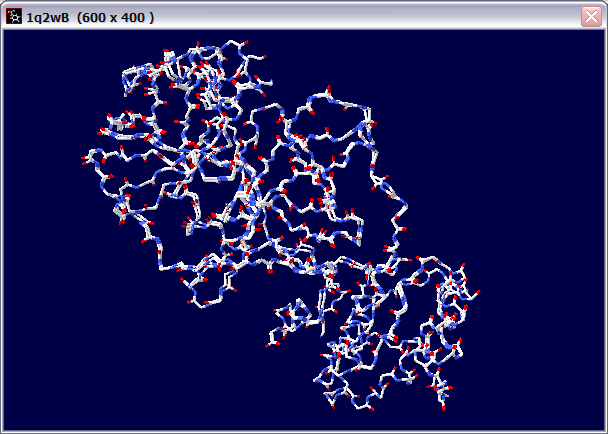
(d) Give the PDB ID for the templet you used.
1. 1Q2WB 1LVOD 1P9UD
(e) Give the Ramachandran plot to verify the quality of your model. How many % of the residues are in the allowed region?
1. Select "Window" "Ramachandran Plot".
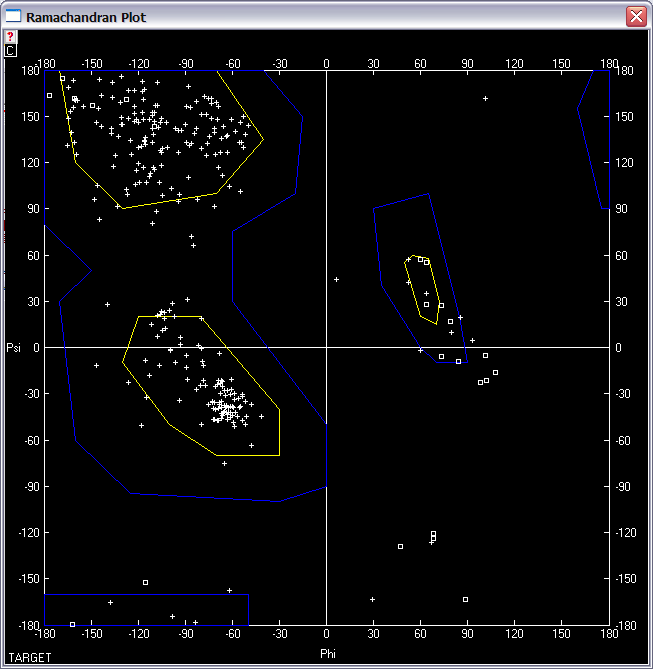
2. Use "Select" "Secondary Structure" "aa
with Phi/Psi out of Allowed Regions", and then select "Window"
"Ramachandran Plot" to check how many residues are out of window.
After counting, there are 18 aa out of allowed regions, and there are total
303 aa in this protein, so it is 94% residues in the allowed region.
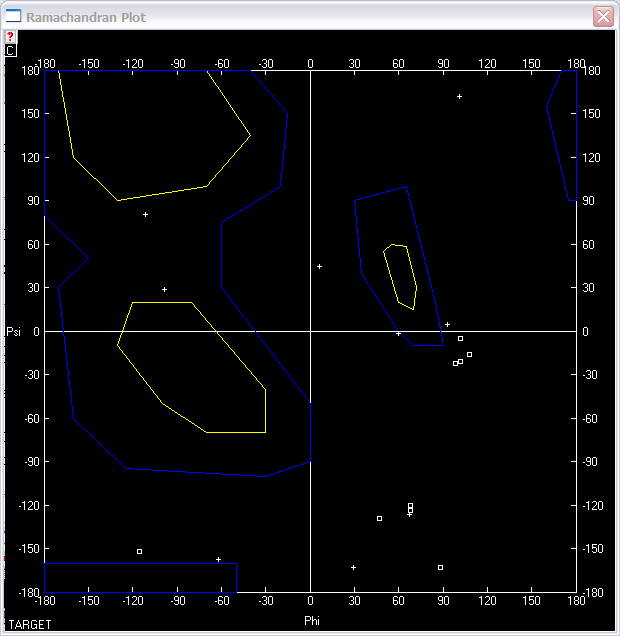
(f) Compare your model with your templet. What is the RMSD of the backbone of the two structures?
1. Select "Fit" "Magic Fit" and choose to fit "backbone atoms only" between the Target and 1q2wB chain.
2. After the fitting, you could see that there are 1176 atoms fitted, and the
RMS was 0.00