Transitions in the visible region of the spectrum are relatively low-energy electronic transitions. Roughly speaking, there are two main kinds of biological structures that have energy levels with spacings in this range: (1) compounds containing certain metal ions (particularly transition metals), and:(2) large aromatic structures and conjugated double-bond systems. The former include some metalloproteins and the latter such conjugated structures as vitamin A. Heme proteins owe most of their intense absorption in the visible region to the conjugated heme group rather than the metal ion. It will be noted that electronic absorption bands are usually quite broad. This is because they actually consist of a large number of closely spaced subbands, each corresponding to a different change in vibrational energy accompanying the change in electronic state. In macromolecules such as proteins additional broadening results : vibrational energy accompanying the change in electronic state ~see from the fact that individual chromophores of a given type may exist in a number of different environments.
In the near-ultraviolet region of the spectrum (200 to 400nm), many biochemicals exhibit strong and useful absorption bands. In keeping with the rough "particle in a box" rule, it is not surprising that these are often associated with small conjugated ring systems, both aromatic and heterocyclic, whereas the large conjugated systems absorb in the visible region. Thus among the amino acids, tyrosine, phenylalanine, and tryptophan absorb most intensely in this region. Most of the absorption of proteins in the 280-nm region results from the presence of these residues. Similarly, all of the purine and pyrimidines bases absorb stronly in the near ultraviolet. Most of these absorption bands appear to correspond to transitions of p electrons in the ring to antibonding p orbitals (the so-called p- p* transitions).
Farther in the ultraviolet, nearly everything absorbs. Suppose we consider due, of course, to the aromatic side chains, but below about 230 nm we begin to encounter not only transitions in other amino acid side chains but also those involving electron displacements in the peptide backbone itself. There appears to be a particularly intense absorption of the latter type at about 190 nm and a weaker one at about 220 nm. These regions will be discussed in circular dichroism (CD).
[Near-ultraviolet absorption bands of some amino acids and nucleotides]
So far, we have made no distinction between the spectra of maciomolecules and those of their monomers. As a first approximation this is justified, especially in the case of random-coil polymeric structures. However, when a definite secondary structure is established in a macromolecule, certain changes are commonly observed in the spectrum. In many cases these have been used as diagnostic tools to detect and follow configurational changes.
Consider, for example, the following effect: When a protein is hydrolyzed to its constituent amino acids, the spectrum of the products is somewhat different from that of the intact protein. Similar changes are observed when the protein is denatured, that is, when its definite secondary and tertiary folding, are disturbed. These observations suggest that the environment of the absorbing residues must be different in the folded protein molecule than when the amino acids are in free contact with the solvent. While the theory is still somewhat incomplete, the effect can be measured with considerable accuracy by difference spectrophotometry. In this technique the optical density difference between the substance substance in two states is measured directly (using, for example, a double-beam spectrophotometer). Thus the change can be used to follow such processes as denaturation.
|
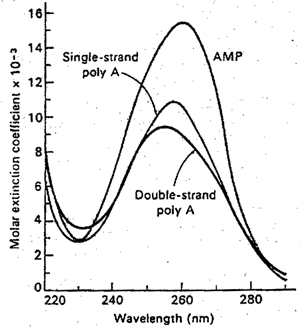
Hypochromism of polyriboadenylic acid. Note the thhe form of the polymer spectrum differs little from that of the monomer but that the intensity of absorption is considerably reduced. |
With nucleic acids even more dramatic effects are observed. The above figure contrasts polyriboadcnylic acid with an equal concentration of its monomer, adenosine monophosphate, while the followinf figure shows the change produced in the spectrum of E. coli DNA by heating to 90oC with accompanying denaturation.
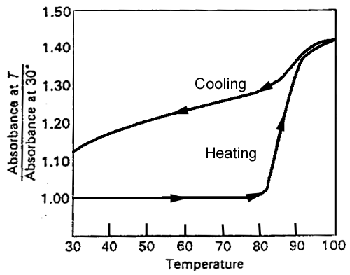 |
The "melting" of DNA as followed by absorption at 260 nm. Note that the sharpness of the transition in cycle 1. This is typical of a cooperative process. Also note that recovery of the ordered structure is not complete on rapid cooling. |
Let us consider the simpler case of the synthetic polyriboadenylic acid. Two effects are observed; the polymer shows a lower absorprion intensity than the monomer (hypochromism), and in the polymer the maximum is shifted to a wavelength almost 3 nm lower.
Both these effects have been explained on the basis of the helical configuration of the nucleotide polymers, in such a regular, closely packed structure the chromophores cannot be considered to be independent of one another. The electronic displacement that occurs when one base absorbs a quantum of energy is felt in the neighboring bases as a modification of the electrical field. If the interaction is strong, it becomes impossible to speak any longer of an absorption "localized" in a particular residue. Rather than the single, N-fold degenerate absorption frequency that a polymer of N independent groups would exhibit, an excilon bond, with N closely spaced but distinct levels, is now observed. The total absorption intensity will not necessarily be distributed equally among the various levels; in this way a shift in the mrtsimum wavelenth of absorption may be produced.
A simple example will illustrate the principle. Suppose we have a dimer of a substance that in the monomeric form has an absorption t the frequency n0. If in the dimer there is interaction of energy U between the chromophores, the single monomer band will be split into two bands at frequencies
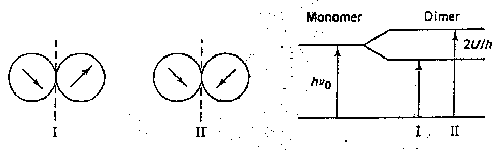 |
n1 = n0 + U/h n2 = n0 - U/h |
The relative absorption intensities will depend on the geometric relation between the transition moments in the two chromophores. In a real system the vibrational broadening of the bands would probably prevent resolution, and we might observe only a shift inn frequency of maximum absorption.
The hypochromism of polymeric structures also arises from intramolecular interactions. Due to interactions with the solvent and surrounding polymer, some of the transitions of the monomer groups may be more favored and others less. Thus the hypbchromism of the 260-nm band in polyadenylic acid or DNA, and its change with conformation, is not surprising. A very,oeneral rule of spectroscopy holds that mere configurational modifications that do not change the fundamental electronic structure should not change the total absorption, integrated over all bands. If this is to hold for the nucleic acids in question, we must postulate an equal hyperchromism somewhere else, presumably in the inaccessible low-wavelength region.
The hypochromism in the ultraviolet bands of the nucleic acids has become an exceedingly useful tool for the observation of configurational change since spectroscopic absorption measurements are rapid and precise.
[BACK]