 Stephen
L. Mayo *
Stephen
L. Mayo *Stephen
L. Mayo *
- the bba motif typified by the zinc finger DNA binding module.
- the second zinc finger module of the DNA binding protein Zif268was selected as design template.
- the amino acids considered at the core positions :
Ala, Val, Leu, Ile, Phe, Tyr, and Trp.
- the amino acids considered at the surface positions :
Ala, Ser, Thr, His, Asp, Asn, Glu, Gln, Lys, and Arg.
- the combined core and surface amino acidsets (16 amino acids) were considered at the boundary positions.
- Two of the residue positions (9 and 27) have f angles greater than 0 degree and are set to Gly by the sequence selection algorithm to minimize backbone strain.



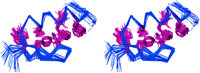
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
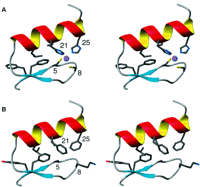
The overall backbone rmsd is 1.98 A for residues 3 to 26 and only 0.98 A for residues 8 to 26. The largest difference between FSD-1 and the target structure occurs from residues 4 to 7, with a displacement of 3.0 to 3.5 A of the backbone atompositions of strand 1.

|
||||||||||||||||||||||||||||||||||
 SA
SA are
the 41 simulated annealing structures,
SA is the average structurebefore
energy minimization, (SA)r is the restrained energy minimizedaverage
structure, and SD is the standard deviation.
are
the 41 simulated annealing structures,
SA is the average structurebefore
energy minimization, (SA)r is the restrained energy minimizedaverage
structure, and SD is the standard deviation.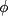 ,
, 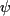 , angular
order parameters (
, angular
order parameters ( (degrees)
(degrees)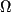 (degrees)
(degrees) coordinates
(residues 15 to 26) and the least-squares plane fit to the C
coordinates
(residues 15 to 26) and the least-squares plane fit to the C