Mass spectrometry (MS) has joined the powerful arsenal of techniques used for structural characterization of biological molecules (1-4). MS can determine the mass of the molecule and the masses of fragments from it, data that are especially valuable for sequencing linear biomolecules. MS's unusual attributes in sampling, sensitivity, speed, simplicity, separation, and specificity have earned this technique multiple uses in the characterization of biomolecules.
Large biomolecules can now be ionized and introduced routinely into MS instruments with the sampling techniques of matrix-assisted laser desorption ionization (MALDI) and electrospray ionization (ESI). In MALDI, laser energy absorbed by the matrix surrounding the biomolecule "explodes" it into the gas phase, whereas in ESI, a sprayed solution of the biomolecule gives electrostatically charged droplets that yield gaseous ions during evaporation. These methods are also unusually sensitive, producing molecular weight (Mr) information for peptides (3, 4) or proteins (5) at the attomole level (10-14 g of a 10-kD molecule). Mass (m) values can be measured over a mass range of >100 kD, using time-of-flight (TOF) and Fourier transform (FT) ion cyclotron resonance (6) spectrometers, with unusual speed (TOF, <1 ms; FT, ~1 s), mass accuracy (TOF, 1/105; FT, 1/106), and resolving power (TOF, 104; FT, 106).
Data Simplicity
MS can be used for sequencing of both proteins and DNA. The order of
the building blocks of a linear molecule can be derived from its mass
plus the mass of its fragmentation
products. For protein fragments, an NH2-terminal Gly is indicated
by the mass corresponding to either
H-Gly or (Mr - H-Gly). Thus masses
of 58.03, (Mr - 129.07), 201.08, and (Mr
- 114.05) daltons indicate the sequence H-Gly-Ala-X-Ser-Pro-OH, based on
the component masses 1.01-57.02-71.04-X-87.03-97.05-17.00.
For the MS fragmentation of proteins, the multiply charged molecular ions
formed by ESI are the most readily dissociated. Alternatively, enzymatic
digestion of the protein produces peptides whose masses
can provide such fragment data. For DNA, dissociation of ESI-produced negative
ions from oligonucleotides as large as 39 kD provides extensive (7)
or, for a 50-nucleotide oligomer DNA (8), complete
sequence information.
Separation and Specificity
Modern MS instrumentation provides a powerful alternative to chromatographic
separation methods. For example, all products of an enzymatic digestion
of a 191-kD protein (9) were introduced by ESI into
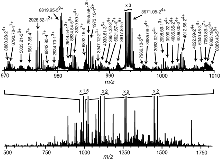
a 9.4-T FTMS (6). In the resulting spectrum (Fig. 1),
each molecular ion is represented by an isotopic peak cluster. An automated
data reduction program (10) locates each cluster,
separates overlaps, and assigns z (charge) and m values to
each; with unusual specificity, Fig. 1 shows 759 clusters corresponding
to 528 mass values as large as 30 kD.
After such a mixture of molecular ions is separated (referred to as MS-I), an ion species of sufficient abundance can be dissociated and its products mass analyzed (MS-II) to provide sequence information (MS/MS). Nanoflow liquid chromatography (LC) coupled by ESI to an ion trap MS/MS can automatically determine Mr values and MS/MS spectra of peptides in the 10 to 50 attomole range (3). This approach has allowed the identification of peptides presented to the immune system in association with major histocompatibility complexes and of melanoma antigens. FTMS provides exact mass data at the 10-attomole level for MS/MS spectra of ionized peptides (3) and proteins (5).
Noncovalent Binding Energies
Accurate thermodynamic values for ESI gaseous ions of noncovalent complexes
can now be determined by black-body infrared dissociation in FTMS. Such
activation energies for the gas phase dissociation of double strand oligonucleotide
anions correlate with the corresponding dimerization enthalpy in solution
(11). ESI/MS may also be useful for rapid screening
of the relative affinities of combinatorially prepared substrates in complex
mixtures (12, 13). However, these
affinities are affected by the absence of aqueous competition; H bonding
becomes much stronger and hydrophobic bonding much weaker (14).
Proteomics
MS has revolutionized the ability to characterize the thousands of
cellular proteins expressed by a genome (4). Separation
and visualization of these proteins by two-dimensional polyacrylamide gel
electrophoresis (2D-PAGE) is well established. However, the micro-Edman
technique for identifying the protein in a 2D gel spot largely has been
replaced by the more sensitive and efficient MS technique of peptide mapping
from in situ spot digestion (4, 15).
MALDI-TOF spectra can be obtained directly from the complex peptide mixture
extracted from the digested protein spot. Usually this accuracy is sufficient
for identification (4) by automated sequence database
searching (16), with only 2 to 5 min required per
sample. For more definitive information, nano-ESI MS/MS of the sample can
provide partial sequence information on the individual peptides that is
sufficient to retrieve a single protein from the database.
These strategies have been used to determine the identities and organization of the 30 major protein components of the 50-MD yeast nuclear pore complex (17), the sole mediator of the macromolecular exchange between the nucleus and the cytoplasm.
Direct MS/MS of protein molecular ions, which avoids the digestion to peptides, can also provide fragment masses for database searching (18), but extraction of larger proteins from 2D gel spots can be difficult (19). Alternatively, 1D separation methods such as LC (3, 15) or capillary electrophoresis (CE) (4, 20, 21) coupled to ESI/MS can be a powerful 2D separation method for identifying proteins in mixtures (5, 21).

Top-Down Protein Characterization
Protein structures predicted from the corresponding DNA sequence can
be incomplete because of posttranslational modifications or DNA sequence
errors. In contrast, "top-down" MS measurement of the masses
of a protein and its fragments (2) can characterize
modifications and errors and locate derivatized active sites (2,
22, 23). An enzyme with a predicted
size of 34 kD and of unknown function yielded an ESI/FTMS spectrum with
two components of Mr = 7310.74 and 26896.5 (22).
Isolation and dissociation of the latter ions showed that the products
indicated a protein (ThiF) of Mr = 26896.1 that matched
the DNA-predicted COOH-terminal sequence. Dissociation of the 7310-dalton
ions (enzyme ThiS) corrected DNA sequence errors and predicted an Mr
of 7310.70. The corrected sequence showed a COOH-terminal Gly-Gly, identical
to that of ubiquitin. Reflecting its enzymatic function, treating ThiFS
with adenosine triphosphate formed the COOH-terminal adenosine monophosphate
adduct, as shown by its correct Mr value; this was converted
to SH by exposure to a sulfur source (verified by a correct Mr
and MS/MS spectrum). This showed that ThiFS plays a key role in the sulfur
insertion forming the thiazole ring of thiamin.
As a further illustration, the ESI/FTMS spectrum of the enzyme, thiaminase, which degrades thiamin, showed Mr values of 42,127, 42,197, and 42,254; none agreed with the DNA prediction (23). However, a pyrimidine suicide substrate that mimics thiamine and binds covalently to the active site increased the Mr value of all three by the expected ~107 daltons, indicating that all three constituents are enzymatically active. Dissociation of the mixed enzyme molecular ions gave fragment ions of 5981.17, 6052.19, and 6109.21 daltons (Dm = 71.02 and 57.02 daltons). Assignment of the remaining fragment ions showed that the components differed by an extra NH2-terminal Ala (71.04 daltons) and Gly (57.02 daltons) and restricted the location of the DNA sequence error that led to the Mr value discrepancies. To localize the enzyme site modified by the suicide substrate, thiaminase was derivatized with an isotopically mixed d0/d3 substrate and digested with Asp-N; selecting the labeled Asp90-Gly122 from the complex ESI/FTMS spectrum was facilitated by its far broader isotopic cluster. MS/MS narrowed the active site location to Pro109-Phe118, while fragment ions consistent with the loss of the substrate label with an attached sulfur atom showed that the only possible labeling site in this 379-residue protein is at Cys113 (23).
De novo Sequencing
The protein MS/MS spectra described above provided only partial sequence
data. In a protein mass spectrum, the
position and mass of an unmodified
or modified amino acid is indicated only if mass
values are produced by cleavages on both sides of the residue. All conventional
ion dissociation methods, such as collisionally activated dissociation
(CAD), cleave the weakest bonds to yield the same mass
products in similar abundance; dissociation of a 10-kD protein ion yields
less than half of the mass values needed
for complete sequencing. However, the bonds cleaved by the new electron
capture dissociation (ECD) method are little affected by their bond dissociation
energy (24), yielding far more extensive cleavages.
For the 76-residue ubiquitin (8.6 kD), for example, mass
data from one CAD and two ECD spectra provide complete sequence information
(Fig. 2) (10). In the future, the dissociation of
larger proteins to pieces of this size, followed by their sequencing and
ordering, may be possible for quantities as small as 10-15 mol.
References
The authors are in the Department of Chemistry and Chemical Biology,
Cornell University, Ithaca, NY 14853-1301, USA.