





Analogous to the reasoning behind saving one member of productive ("interesting") families to prevent excessive minimization within a binding mode, it is desirable to do the same for non-productive (bumping) families. Any orientation found to exceed the number of allowed bumps constitutes a family within which no other orientations will be permitted (subject to the degenerate_save_interval parameter, see below), thus saving precious minimization time in uninteresting binding modes.
Caution: Degeneracy checking is still an experimental feature. It is designed to be used as a tool in speeding up DOCK runs involving force-field score minimization, but it can often be equally effective to use minimization without degeneracy checking with greatly reduced bin sizes.
Theory
Problem description
The tricky part of the problem is that we have to know where in the active
site an orientation lies based solely on the sphere-atom pairings involved in
the match; at this stage in the program we have no information of atom
coordinates as the ligand has not yet been placed into the context of the
receptor. Furthermore, we have to know when the same geometry has been
produced with different pairings. Consider the simple model depicted
below, with E being the "receptor," F the "ligand," and circled
points are spheres. Using a three-node match, we can superimpose F onto
E by the pairings b3, c4, d5. However, the matching of a2, e6, d5
produces the identical geometric orientation. Given the latter pairing, we
must be able to recognize that this will generate an orientation degenerate
with the former.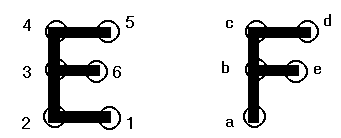
Degeneracy check
When a unique orientation is found (e.g. the very first match), the
program saves the nearest sphere to every atom in the ligand - this
information is, of course, dependent on the orientation relative to the
receptor. Every subsequent orientation must be checked for degeneracy,
i.e. has this geometry been seen before? We may only use the four
sphere-atom pairings used to generate the match, otherwise we waste the time
required to orient the orientation in the site and transform the
coordinates (for sake of discussion, we will assume four nodes are required for
a match). A simple check to see if all four pairings occurred simultaneously
in a previous unique match tells us the answer. Note that we must save the
nearest sphere to every atom in the ligand for unique matches to allow
detection of similar geometries produced by different pairings.
Information storage
Saving a list of all matches for each possible sphere-atom pairing
(maxlig (500) by maxpts (120) by at least 10,000 unique matches
at 4 bytes per integer = 2.4Gb array with current dimensions) requires a
tremendous amount of memory, since Fortran does not support dynamic memory
allocation. Moreover, much of this array would be empty, so this is a problem
that lends itself to hashing. We employ open addressing with double hashing,
as per Knuth. The hash table allows us to store only non-zero elements of the
3-dimensional array mentioned earlier, with clever methods of retrieving
information given a hash code. The hash code is a function of one sphere-atom
pairing and dictates where in the table matches containing this pair have be
found. Thus, given one sphere-atom pairing, one can quickly retrieve all other
orientations which contained this pairing.
Sensitivity reduction
All that is required for differentiating orientations is a small set of way
points in the active site. Here, a way point is merely a geometric descriptor
which signals the occupancy by the ligand of a particular portion of the active
site volume. A typical active site will be represented by on the order of
50-100 spheres, usually overkill for such a simple task. Each way point
describes a particular volume within the site, this volume inversely
proportional to the number of way points. A ligand orientation is described by
the way points its atoms "see." The more way points one uses, the more
discerning the program will be in differentiating active site volume.
Rephrased, fewer degenerate orientations will be removed because more matches
will be considered unique. The goal here is the opposite: to reduce
the number of orientations passed through to the minimizer. We can easily
reduce the number of way points by clustering the sphere set ("true spheres")
to generate a reduced set of virtual spheres. All neighboring true
spheres are jointly averaged into one virtual sphere. This creates an even
distribution of way points throughout docking space.
Degeneracy stringency
Another method for removing more degenerate orientations is to allow some slop
in comparing the sphere-atom pairings with those in unique matches. This
feature is termed "wobble," as we may permit a non-zero number of "mistakes" in
the degeneracy check. The predictable effect of introducing wobble is to
increase the number of degenerate orientations because binding modes are
smeared out over a larger volume.
Safety net
Because the first orientation in a family is deemed the representative of a
particular binding mode, our depiction of this binding mode is highly dependent
on the quality of the orientation. All future orientations in this family will
be considered degenerate to the initial member. If the quality (e.g.
force-field score) is very poor, then we unfairly represent this binding mode.
It would be beneficial to afford popular binding modes renewed chances at
locating an optimal representative. The parameter
degenerate_save_interval dictates how often we must find a degenerate
orientation in a family before orienting and minimizing another member. The
degenerate_save_interval parameter has the desirable effect of smoothing
our sampling over all binding modes.Usage
Virtual spheres are a reduced set of averaged spheres derived from your
sphgen sphere center output used in determining orientation degeneracy.
These spheres act as way points within the active site for defining the
geometry of an orientation. The following program works by clustering all
spheres which have any neighbor within a specified radius and computing an
average position for a so-called "virtual sphere." The fewer virtual spheres
used the more orientations will be considered degenerate and run time will be
shorter; but beware, too few virtual spheres results in overlooked binding
modes.
Create virtual spheres as follows. Reduce the actual sphgen sphere
cluster to virtual spheres by running the interactive program
virtual_spheres on your sphgen sphere center file. For the
intersphere cutoff distance (parameter vsph), use a value of 2.0
Å. Smaller cutoff distances have less of a reducing effect on the
original sphere set than do larger cutoff distances. Having more virtual
spheres (smaller cutoff distance) provides fewer degenerate orientations.
Consequently, runtime will be longer but accuracy improved. Note that a file
called merge.lst was created - this file lists which true sphgen spheres
were averaged (merged) into which virtual sphere. View the virtual sphere PDB
file on a graphics terminal to verify that they are evenly spaced at a
sufficient density for your application.
.sph extension will be written.
Add the check_degeneracy keyword to your INDOCK parameter file. Other
parameters should be customized by using the appropriate keyword, as described
below:
OUTDOCK file to insure that the run is proceeding or has completed
successfully. Confirmation that minimization and degeneracy checking have
taken effect should be reported. A number of statistics concerning degeneracy
and minimization are given at the end of the OUTDOCK file.
SINGLE runs: For each center for which matching has been completed,
statistics on the number of matches attempted, number of unique orientations
found, and number and percent of degenerate orientations are given. A file
called family.log is also written which contains an orientation degeneracy
histogram, providing the energy, starting energy, and rms deviation of each
unique orientation, as well as how many times it was seen. Also included are
the parent orientation for any children saved due to the
degenerate_save_interval parameter.
SEARCH runs: For each compound, the number of matches attempted, number of unique matches, and number of degenerate orientations are reported.
An interesting feature: the family.log statistics (energy, starting energy,
and RMS deviation) may actually be compiled while still minimizing every
orientation generated. This can be accomplished by setting
degenerate_save_interval to 1 (thus saving every multiple in a family)
and turning check_degenerate_children off (degeneracy_wobble may
assume any value). This method does, of course, require the creation of all
files required for degeneracy checking.
Cautions
Memory requirements
Degeneracy checking can be very memory intensive because of all the
information that must be stored. Should memory problems be encountered, the
easiest work-around is to reduce the dimensions of the arrays in the header
file degeneracy.h. Specifically, maxfamily may be reduced to the
maximum number of expected unique matches - I'd recommend going no lower than
5,000. Also, several alternative prime number pairs are given in this header
file for dimensioning the hash table with the maxhash and hash2
parameters.Filling of hash table
The hash table used in degeneracy checking is an effective tool for
circumventing the immense memory requirements for the task at hand. With
standard DOCK runs, using a hash table can be just as rapid as storing
all the required information in memory, were that much memory available.
However, when the hash table becomes full, performance can degrade very
rapidly. It is therefore imperative that the hash table be dimensioned
sufficiently large. Degeneracy checking is not meant to be used for
long SINGLE mode dock runs which generate upwards of hundreds of thousands of
matches. The OUTDOCK file reports the percentage of the hash table used in
single mode runs - this number should probably be less than 50% for efficient
runs. Also, an "average number of search steps per match" (supplied at the end
of the OUTDOCK file) greater than 5 can indicate performance degradation due to
a full hash table.Focusing
It has not yet been established if degeneracy checking is compatible with
focusing. Bear in mind that focusing is a form of discrete optimization, while
minimization is a continuous optimization. It may not be efficient, nor
desirable, to use both concurrently.
Curator: Daniel Gschwend, gschwend@cgl.ucsf.edu (rev. 1 September 1995)