





where each term is a double sum over ligand atoms i and receptor atoms j, Aij and Bij are van der Waals repulsion and attraction parameters, rij is the distance between atoms i and j, qi and qj are the point charges on atoms i and j, D is the dielectric function, and 332.0 is a factor that converts the electrostatic energy into kilocalories per mole.(1)
An efficient grid calculation requires the use of a geometric mean approximation for the van der Waals parameters:
where the single-atom-type parameters are calculated from van der Waals radius, R, and well depth, epsilon, according to:and
(2)
Using this approximation, eq. 1 can be rewritten:and
(3)
Three values are stored for every grid point k, each a sum over receptor atoms that are within a user-defined cutoff distance of the point:(4)
These values, with or without interpolation, are multiplied by the appropriate ligand values to give the interaction energy. chemgrid calculates the grid values and stores them in files. The values will be read in during a DOCK run and used for force field scoring. Substituting eq. 5 into eq. 4, the interaction energy is: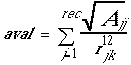
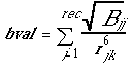
(5)
Atoms that fall outside the grid, if any, are given interaction energies of zero.(6)
The user determines the location and dimensions of the grid box. It is not necessary for the whole receptor to be enclosed; only the regions where ligand atoms may be placed need to be included. The box merely delimits the space where grid points are located, and does not cause receptor atoms to be excluded from the calculation. Besides a direct specification of coordinates, there is an option to center the grid at a sphere cluster center of mass. Any combination of spacing and x, y, and z extents may be used as long as the total number of points does not exceed the array size, maxgrd. The PDB box file from the program showbox is used for setting up the grid location. The programs showesp and showprobe may be helpful for examining the output grid files.
Input
The input file names and parameters are read from a file called INCHEM, which should not contain any blank lines:
variable name Fortran format example recfil A80 4dfr_model.pdbtable A80 prot.table.ambcrg.ambHvdwfil A80 vdw.parms.ambinbox A80 4dfr.boxgrddiv F 0.30estype I 1esfact F 4cutoff F 10.0pcon ccon 2F 2.3 2.8grdfil A80 4dfr_chem
prot.table.ambcrg.ambH
(protein, AMBER united-atom charges, AMBER hydrogen names),
prot.table.ambcrg.pdbH (protein, AMBER united-atom charges, PDB hydrogen
names), and na.table.ambcrg (nucleic acid, AMBER united-atom charges). The
united-atom parameters are from the 1984 AMBER reference (Weiner et al.,
1984). Each table consists of comment lines and atom parameter lines:
! receptor parameter file sample
! '!' at the beginning of a line indicates a comment
!
!aaaxxxrrrnnnncqqqqqqqqxtt format (A4, 3X, A3, A4, A1, F8.3, X, I2)
! atom name, residue name, residue number (read as characters), chain
! indicator, charge, van der Waals type
!
N ALA -0.520 8 ALANINE
CA ALA 0.215 4
C ALA 0.526 1
O ALA -0.500 11
H ALA 0.248 6
CB ALA 0.031 2
Receptor atom parameterization is done by hashing and typically takes only a
few seconds. In the table, the atom name is mandatory, as well as the charge
and van der Waals type. The residue name, residue number, and chain indicator
are optional. A receptor atom will be parameterized according to most specific
match that can be found. The order of lines in the table is immaterial. The
user may add to the table to accommodate nonstandard residues, or may create a
table with completely different parameters. vdw.parms.amb
is located in the parms subdirectory and contains AMBER
all-atom and united-atom parameters (Weiner et al., 1984; Weiner et
al., 1986). This table consists of comment lines and lines containing
SQRT(A) and SQRT(B) for each van der Waals type. The lines must be in order of type, without any
skipped integers. The types are referred to by number in the receptor parameter
file, table.REMARK lines of this PDB-format file; the
remaining lines are ignored. The dimensions are aligned along the x,
y, and z axes.Output
Three output files are generated in addition to the grid files and the PDB box
file: OUTCHEM, OUTPARM, and PDBPARM. OUTCHEM lists the parameters specified
in INCHEM, so that one can verify whether the intended values have been read
in. PDBPARM explicitly lists the parameters along with the coordinates of each
receptor atom. Whenever parameters are not found for a given receptor atom,
they are set to zero (no volume and no charge), and a message is written to
OUTPARM. OUTPARM ends with the calculated net charge on the receptor molecule.
It is a good idea to check the net charge and make sure that it is correct (it
should be an integer!) and agrees with that value from the accessory program
charge. For convenience in error detection, a line is written for each
residue calculated to have a net charge; while +1 and -1 are normal, a
noninteger charge probably indicates a parameterization error for that residue.
The lines in OUTPARM reporting charged residues each contain the string
"CHARGED RESIDUE".
 What's all this stuff in my OUTPARM file?
What's all this stuff in my OUTPARM file?
Sample Output
receptor pdb file:
4dfr_model.pdb
receptor parameters will be read from:
/usr/einstein/dock/parms/prot.table.ambcrg.ambH
van der Waals parameter file:
/usr/einstein/dock/parms/vdw.parms.amb
input box file defining grid location:
4dfr30.box
box center coordinates [x y z]:
25.00000 68.00000 46.00000
box x-dimension = 25.00000
box y-dimension = 15.00000
box z-dimension = 25.00000
grid spacing in Ångstroms
0.3000000
grid points per side [x y z]:
85 51 85
total number of grid points = 368475
a distance-dependent dielectric will be used
the dielectric function will be multiplied by 4.00
cutoff distance for energy calculations:
10.00000
distances defining bumps with receptor atoms:
receptor polar atoms 2.30
receptor carbon atoms 2.80
output grid prefix name:
test
Curator: Daniel Gschwend, gschwend@cgl.ucsf.edu (rev. 1 September 1995)