Overview
grid creates the grid files necessary for rapid score evaluation in dock. Three types of scoring are available: contact, chemical and energy scoring. The scoring grids are stored in files ending in *.cnt, *.chm, and *.nrg respectively. When docking, each scoring function is applied independent of the others and the results are written to separate output files.
grid also computes a bump grid which identifies whether a ligand atom is in severe steric overlap with a receptor atom. The bump grid is identified with a *.bmp file extension. The file containing the bump grid also stores the size, position and grid spacing of all the grids.
The grid calculation must be performed prior to docking. The calculation can take up to 45 minutes, but needs to be done only once for each receptor site. Since dock can perform continuum scoring without a grid, the grid calculation is not always required. However, for most docking tasks, such as when multiple binding modes for a molecule or multiple molecules are considered, it will become more time efficient to precompute the scoring grids.
Bump Checking
Prior to scoring, each orientation can be processed with the bump filter to reject ones that penetrate deep into the receptor. Orientations that pass the bump filter are then scored and/or minimized with any of the available scoring functions.
The way a bump is detected has been changed from previous versions of dock. Instead of a strict distance cutoff, a bump is based on the sum of the van der Waals radii of the two interacting atoms. The user specifies what fraction of the sum is considered a bump. For example, the default definition of a bump is if any two atoms approach closer than 0.75 of the sum of their radii.
The way bump information is stored on a grid has also changed. Rather than storing a True/False value at every grid point, grid stores an atomic radius which corresponds to smallest radius of ligand atom at the grid position which would still trigger a bump. During docking, for a given orientation, the position of each atom is checked with the bump grid. If the radius of the atom is greater than or equal to the radius stored in the bump grid, then the atom triggers a bump. To conserve disk space, the atom radius is multiplied by 10 and converted to a short unsigned integer, which takes up as much memory (1 byte) as the True/False value used in the previous implementation.
Energy scoring
The energy scoring component of dock is based on the implementation of force field scoring (See
20
in the
References
section of the
Users Guide
) in earlier versions of dock. Force field scores are approximate molecular mechanics interaction energies, consisting of van der Waals and electrostatic components:
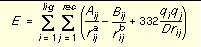 Equation 1
Equation 1
where each term is a double sum over ligand atoms i and receptor atoms j, which include the quantities listed below.
|
E
|
Intermolecular interaction energy
|
|
rij
|
Distance between atoms i and j
|
|
Aij and Bij
|
van der Waals repulsion and attraction parameters
|
|
a and b
|
van der Waals repulsion and attraction exponents
|
|
qi and qj
|
Point charges on atoms i and j
|
|
D
|
Dielectric function
|
|
332
|
Factor to convert electrostatic energy to kcal/mol.
|
Generalization of the VDW component
The van der Waals component of the scoring function has been generalized to handle any combination of repulsive and attractive exponents (providing that a > b). The user may choose to "soften" the potential by using a 6-9 Lennard-Jones function. The general form of the van der Waals interaction between two identical atoms is presented in
Equation 2
,
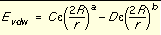 Equation 2
Equation 2
which includes the following additional quantities.
|
C and D
|
Coefficients to be determined
|
|
e
|
Well depth of interaction energy
|
|
R
|
van der Waals radius of atoms
|
The coefficients C and D can be determined given the two following boundary conditions.
 , at
, at 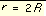 Equation 3
Equation 3
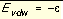 , at
, at 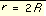 Equation 4
Equation 4
Application of these boundary conditions to
Equation 2
yields an expression of the van der Waals interaction with a generalized Lennard-Jones potential.
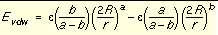 Equation 5
Equation 5
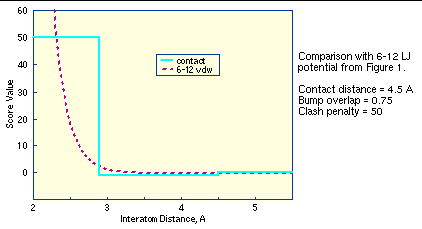
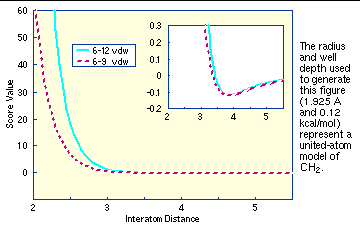
The consequence of using a different exponent for the repulsive term are illustrated in
Figure 1
. Notice that the well position and depth are unchanged, but that the repulsive barrier has shrunk by about a quarter Angstrom.
Precomputing potentials on a grid
By inspection of
Equation 1
and
Equation 5
, the repulsion and attraction parameters (Aij and Bij) for the interactions of identical atoms can be derived from the van der Waals radius, R, and the well depth, e.
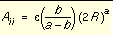 and
and 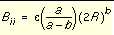 Equation 6
Equation 6
In order to evaluate the interaction energy quickly, the van der Waals and electrostatic potentials are precomputed for the receptor and stored on a grid of points containing the docking site. Precomputing the van der Waals potential requires the use of a geometric mean approximation for the A and B terms, as shown in
Equation 7
.
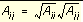 and
and 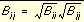 Equation 7
Equation 7
Using this approximation,
Equation 1
can be rewritten:
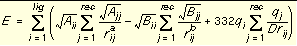 Equation 8
Equation 8
Three values are stored for every grid point k, each a sum over receptor atoms that are within a user-defined cutoff distance of the point:
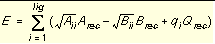 Equation 10
Equation 10
Atoms that fall outside the grid, if any, are given interaction energies of zero.
The user determines the location and dimensions of the grid box using the program
showbox. It is not necessary for the whole receptor to be enclosed; only the regions where ligand atoms may be placed need to be included. The box merely delimits the space where grid points are located, and does not cause receptor atoms to be excluded from the calculation. Besides a direct specification of coordinates, there is an option to center the grid at a sphere cluster center of mass. Any combination of spacing and x, y, and z extents may be used.
Contact Scoring
Contact scoring in grid incorporates the scoring performed with the distmap program developed by Shoichet and Bodian. The score is a summation of the heavy atom contacts (every atom except hydrogen) between the ligand and receptor. A contact is defined as an approach of two atoms within some cutoff distance (usually 4.5 Angstroms). If the two atoms approach close enough to bump (as identified with the bump grid) then the interaction can be penalized by an amount specified by the user. The distance dependence of the contact score is represented in
Figure 2
.
Unlike the convention in distmap, an attractive score in grid is negative and a repulsive score is positive. This switch of sign was necessary to allow the same minimization protocol to be used for contact scoring as implemented for energy scoring.
Chemical Scoring
Chemical scoring is an optional scoring function which is based on energy scoring, but incorporates empirical concepts of molecular interaction. It includes the van der Waals and electrostatic terms from energy scoring, but the attractive portion of the van der Waals term has been modified. The attractive portion is scaled depending on the chemical nature of the interacting atoms.
The philosophy of chemical scoring is to give some control over scoring to the user without requiring any programming effort. Please see the section titled
Chemical Score on page 40
for more discussion.
Chemical labeling of atoms
Atoms are identified with particular chemical labels depending on their atom type and the types of adjacent atoms. See
chem.defn on page 106
for an example of chemical labels and definition rules. The file containing these rules is a text file and can be modified by the user. Consequently, the user can create any set of labels and definitions desired.
Scaling of interactions
The van der Waals interaction between two atoms is scaled based on the rules contained in a user-supplied file (see
chem_score.tbl on page 108
). The scaling factors provided with the distributed version of dock are merely suggestions. They are not derived from first principles, nor are they fitted to experimental data. They should be considered preliminary and can be adjusted depending on the interests of the user and the considerations of a particular test system.
© UC Regents 1998
Top / Up / Previous / Next
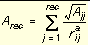 ,
, 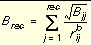 , and
, and 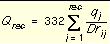 Equation 9
Equation 9