Programs
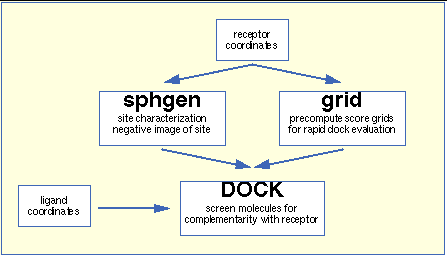
The relationship between the main programs in the dock suite is depicted in
Figure 1.
These routines will be described below; more details can be found in various papers. We list a small subset of papers. Review articles on the method can be found in Kuntz [
1
] and Kuntz, Meng and Shoichet [
2
].
Program
sphgen
identifies the active site, and other sites of interest, and generates the sphere centers which fill the site. It has been described in the original paper: Kuntz, et al. [
3
]. Program
grid
generates the scoring grids; details can be found in Shoichet, Bodian and Kuntz [
4
] and Meng, Shoichet and Kuntz [
5
]. Within the dock suite of programs, the program dock matches spheres (generated by
sphgen
) with ligand atoms and uses scoring grids (from
grid
) to evaluate ligand orientations; descriptions can be found in Kuntz, et al. [
3
] and Shoichet, Bodian and Kuntz [
4
]. Program dock also minimizes energy based scores; description of minimization can be found in Meng, Gschwend, Blaney and Kuntz [
6
].
Several stand-alone docking-related programs exist. Program
cluster
generates alternative clusters of sphere centers within the active site. It uses as input, files from program
sphgen
. For energy scoring, ligand atom van der Waal attractive and dispersive factors and partial charges are also required.
General Concepts
This document is intended to give an overview of the ideas which form the basis of the dock suite of programs. It is not intended to be a reference manual or a user's guide for the programs, nor a substitute for all the papers written on dock. Rather, it gives a synopsis of the structure of the programs and concepts underlying the programs.
The dock suite of programs is designed to find favorable orientations of a ligand in a "receptor." It can be subdivided into (i) those programs related directly to docking of ligands and (ii) accessory programs. We limit the discussion in this section to only those programs and methods related to docking a ligand in a receptor. A typical receptor might be an enzyme with a well-defined active site, though any macromolecule may be used (e.g. a structural protein, a nucleic acid strand, a "true" receptor). We'll use an enzyme as an example in the rest of this discussion.
The starting point of all docking calculations is generally the crystal structure of an enzyme from an enzyme-ligand complex. The ligand structure may be taken from the crystal structure of the enzyme-ligand complex or from a database of compounds, such as the
Cambridge Crystallographic Database
[
7
] or the Concord-generated [
8
] set of coordinates from the
Available Chemicals Directory
, or ACD, (from Molecular Design, Ltd., San Leandro, CA). The primary consideration in the design of our docking programs has been to develop methods which are both rapid and reasonably accurate. These programs can be separated functionally into roughly two parts, each somewhat independent of the other:
-
Routines which determine the orientation of a ligand relative to the receptor.
-
Routines which evaluate (score) a ligand orientation.
There is a lot of flexibility. You can generate orientations outside of dock and score them with the dock evaluation functions. Alternatively, you can develop your own scoring routines to replace the functions supplied with dock.
The ligand orientation in a receptor site is broken down into a series of steps, in different programs. First, a potential site of interest on the receptor is identified. (Often, the active site is the site of interest and is known a priori.) Within this site, points are identified where ligand atoms may be located. A routine from the dock suite of programs identifies these points, called sphere centers, by generating a set of overlapping spheres which fill the site. Rather than using dock to generate these sphere centers, important positions within the active site may be identified by some other mechanism and used by dock as sphere centers. For example, the positions of atoms from the bound ligand may be used as these sphere centers. Or, a grid may be generated within the site and each grid point may be considered as a sphere center. Our sphere centers, however, attempt to capture shape characteristics of the active site (or site of interest) with a minimum number of points and without the bias of previously known ligand binding modes.
To orient a ligand within the active site, some of the sphere centers are "matched" with ligand atoms. That is, a sphere center is "paired" with an ligand atom. Many sets of these atom-sphere pairs are generated, each set containing only a small number of sphere-atom pairs. In order to limit the number of possible sets of atom-sphere pairs, a longest distance heuristic is used; (long) inter-sphere distances are roughly equal to the corresponding (long) inter-atomic ligand distances. A set of atom-sphere pairs is used to calculate an orientation of the ligand within the site of interest. The set of sphere-atom pairs which are used to generate an orientation is often referred to as a match. The translation vector and rotation matrix which minimizes the rmsd of (transformed) ligand atoms and matching sphere centers of the sphere-atom set are calculated and used to orient the entire ligand within the active site.
The orientation of the ligand is evaluated with a shape scoring function and/or a function approximating the ligand-enzyme binding energy. All evaluations are done on (scoring) grids in order to minimize the overall computational time. At each grid point, the enzyme contributions to the score are stored. That is, receptor contributions to the score, potentially repetitive and time consuming, are calculated only once; the appropriate terms are then simply fetched from memory.
The shape scoring function is an empirical function resembling the van der Waal attractive energy. To generate the shape score, the receptor terms from the grid point nearest to each non-hydrogen ligand atom are summed together. That is, the shape score is determined simply by the position of each ligand atom on the shape scoring grid.
The ligand-enzyme binding energy is taken to be approximately the sum of the van der Waal attractive, van der Waal dispersive, and Coulombic electrostatic energies. Approximations are made to the usual molecular mechanics attractive and dispersive terms for use on a grid. To generate the energy score, the ligand atom terms are combined with the receptor terms from the nearest grid point, or combined with receptor terms from a "virtual" grid point with interpolated receptor values. The score is the sum of over all ligand atoms for these combined terms. In this case, the energy score is determined by both ligand atom types and ligand atom positions on the energy grids.
As a final step, in the energy scoring scheme, the orientation of the ligand may be varied slightly to minimize the energy score. That is, after the initial orientation and evaluation (scoring) of the ligand, a grid-based rigid body simplex minimization is used to locate the nearest local energy minimum. The sphere centers themselves are simply approximations to possible atom locations; the orientations generated by the sphere-atom pairing, although reasonable, may not be minimal in energy.
Specific Concepts: mechanisms to limit cpu time
Sphere Centers
From an unknown source: "...what's good about dock is that it uses spheres; what's bad about dock is that it uses spheres..."
Spheres are generated to fill the target site. The sphere centers are putative ligand atom positions. Their use is an attempt to limit the enormous number of possible orientations within the active site. Like ligand atoms, these spheres touch the surface of the molecule and do not intersect the molecule. The spheres are allowed to intersect other spheres; i.e. they have volumes which overlap. Each sphere is represented by the coordinates of its center and its radius. Only the coordinates of the sphere centers are used to orient ligands within the active site (see above). Sphere radii are used in clustering.
The number of orientations of the ligand in free space is vast. The number of orientations possible from all sets of sphere-atom pairings is smaller but still large and cannot be generated and evaluated (scored) in a reasonable length of time. Consequently, various filters are used to eliminate from consideration, before evaluation, sets of sphere-atoms pairs, which will generate poorly scoring orientations. That is, only a small subset of the number of possible ligand orientations are actually generated and scored. The distance tolerance is one filter. Sphere "coloring" and identification of "critical" spheres are other filters.
Sphere-sphere distances are compared to atom-atom distances. Sets of sphere-atom pairs are generated in the following manner: sphere i is paired with atom I if and only if for every sphere j in the set and for every atom J in the set,
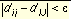 Equation 1
Equation 1
where dij is the distance between sphere i and sphere j, dIJ is the distance between atom I and atom J, and e is a somewhat small user-defined value.
*Note: since dock matches spheres with ligand atoms by comparing distances between sphere pairs and ligand atom pairs, the mirror image of the ligand atoms (used in the match) may be a better fit, in the rmsd sense, to the spheres, than the atoms of the real, non-mirror-reflected ligand. Consider, for example, the distances between the four atoms bonded to a chiral carbon center and the distances between the four atoms bonded to the mirror-image of that chiral carbon center: the distances are the same, but the two sets of four atoms cannot be superimposed upon one another, unless the chirality of one is reversed. In a similar manner, the chirality of the ligand atoms used in the match may be opposite to that of the matching spheres.
Chemical Matching
dock spheres are generated without regard to the chemical properties of the nearby receptor atoms. Sphere "chemical matching" or "coloring" associates a chemical property to spheres and a sphere of one "color" can only be matched with a ligand atom of complementary color. These chemical properties may be things such as "hydrogen-bond donor," "hydrogen-bond acceptor," "hydrophobe," "electro-positive," "electro-negative," "neutral," etc. Neither the colors themselves, nor the complementarity of the colors, are determined by the dock suite of programs; dock simply uses these labels. With the inclusion of coloring, only ligand atoms with the appropriate chemical properties are matched to the complementary colored spheres. It is probably more likely, then, that the orientation generated will produce a favorable score. Conversely, by excluding colored spheres from pairing with certain ligand atoms, the number of (probably) unfavorable orientations which are generated and evaluated can be reduced. Note that requiring complementarity in matching does not mean that all ligand atoms will lie in chemically complementary regions of the enzyme. Rather, only those ligand atoms, when paired with a colored sphere which is part of the sphere-atom match, will be guaranteed to be in the chemically complementary region of the enzyme (provided chirality of the spheres is the same as that of the matching ligand atoms).
Critical Points
The "critical point" filter requires that certain spheres be part of the set of sphere-atom pairs used to orient the ligand [
9
]. Designating spheres as critical points forces the ligand to have at least one atom in that area of the enzyme, where that sphere is located. This filter may be useful, for example, when it is known that a ligand must occupy a particular area of an active site. This filter removes from consideration any orientation that does not guarantee at least one ligand atom in critical areas of the enzyme (provided chirality of the spheres is the same as that of the matching ligand atom).
Scoring Filters
After a ligand is oriented within the active site, the orientation is evaluated. In an attempt to reduce the total computational time, after the ligand is oriented in the site, ligand atoms are first checked to determine whether or not they occupy space already occupied by the receptor. This is often referred to as "bump checking." If too many of such "bumps" are found, then the ligand is likely to intersect the receptor even after minimization; consequently, the ligand orientation is discarded before evaluation.