Chemical Screen
The chemical screen option enables rapid screening of molecules either prior to or following docking. It is requested with the
chemical_screen
parameter. It can be used to perform a
Pharmacophore Screen
or a
Similarity Screen
. It is based on the same chemical labels and definitions as
Chemical Matching
and the
Chemical Score
(see
chem.defn on page 106
). These tools should be viewed as rudimentary in functionality compared to what is available in commercial small-molecule software packages. However, considering their ease of use and compatibility with dock, they should be of interest to the dock user.
The chemical search key encodes information about the number of distances between chemical groups in a molecule and the magnitudes of the distances. The number of distances between two different chemical groups, 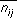 , records information about the composition of the molecule. The distances themselves are stored in a binary fingerprint,
, records information about the composition of the molecule. The distances themselves are stored in a binary fingerprint, 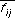 , which records information about the spatial distribution of properties in the molecule. Since the binary fingerprint may lose information about the frequency of occurrence of certain distances, particularly for flexible molecules, the number of distances record is necessary.
, which records information about the spatial distribution of properties in the molecule. Since the binary fingerprint may lose information about the frequency of occurrence of certain distances, particularly for flexible molecules, the number of distances record is necessary.
Keying a database is performed with the
construct_screen
parameter. The fingerprint architecture is specified with the
distance_begin
,
distance_end
, and
distance_interval
parameters. When keying the database, a
Simultaneous Search
of conformations should be performed to generate an ensemble of conformers. For each conformer, the distances between chemical groups is computed and stored in the binary fingerprint. The keyed database is actually a
PTR format
database file with the additional fields for chemical keys. Once the database has been keyed, it may then be searched using the
screen_ligands
parameter.
Pharmacophore Screen
A pharmacophore search is a useful way to prescreen a database to identify molecules that might interact favorably with a small number of receptor atoms with well-defined geometry. It is requested with the
pharmacophore_screen
parameter. The pharmacophore is actually a set of points with associated chemical labels serving as a three dimensional hypothesis for a binding model. The pharmacophore could be constructed from a set of active ligands, or extracted from a receptor structure. This feature is useful when only a few interactions with the receptor are of interest and can be used to construct a pharmacophore. The chemical screen would then be able to discard all molecules that would never be able to make the desired interactions.
Given a molecule a and a pharmacophore b, the rules for determining whether the molecule might satisfy the pharmacophore are for all i and j not equal to i:
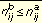 Equation 5
Equation 5
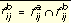 Equation 6
Equation 6
where 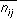 and
and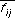 are defined in the
Chemical Screen
section, above.
are defined in the
Chemical Screen
section, above.
In order to account for uncertainty in the distances, a
distance_tolerance
is used to blur the distance key of the molecule.
After the chemical screen run has discarded molecules that could never satisfy the pharmacophore, then a regular dock run can be performed to find the molecules that do satisfy it. In the follow-up run, use a
Simultaneous Search
of conformations along with
Chemical Matching
to the pharmacophore site points.
Similarity Screen
A similarity search is useful to identify all molecules in a database which might be similar in chemical property distribution to a particular molecule of interest. Such a search is useful after docking, once a set of active molecules has been identified. It is activated with the
similarity_screen
parameter. The cutoff for writing out hits is specified with the
dissimilarity_maximum
parameter. Dissimilarity is used to be consistent with the other scoring functions in which smaller values represent favorable values.
Since the chemical key contains both a binary fingerprint and an integer count of distances, a modified Tanamoto index is used as shown in
Equation 7
. This similarity metric maintains the connection between the fingerprint and the count of chemical distances.
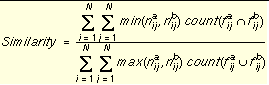 Equation 7
Equation 7
For flexible molecules, the distance fingerprints can become saturated. The similarity can still be discerned by the distance count element of the key.
Some chemical groups may be treated as equivalent to other groups (e.g. hydroxyls and hydrogen bond donors). Such chemical equivalency can be supplied in an editable file (see
chem_screen.tbl on page 109
) and identified with the
chemical_screen_file
parameter.
© UC Regents 1998
Top / Up / Previous / Next