Conformation Search
The conformation of a flexible molecule may be searched or relaxed using the
flexible_ligand
option. Only the torsion angles are modified, not the bond lengths or angles. Therefore, the input geometry of the molecule needs to be of good quality. A structure generated by CONCORD is sufficient.
The user may request a conformation search using the
torsion_drive
parameter and/or torsion minimization using the
torsion_minimize
parameter. The torsion angle positions reside in an editable file (see
flex_drive.tbl on page 111
) which is identified with the
flex_drive_file
parameter. Internal clashes are detected during the torsion drive search based on the
clash_overlap
parameter, which is independent of scoring function.
If
multiple_ligands
are being processed, then the
flexible_bond_maximum
cutoff is used to discard overly flexible molecules.
When scoring is requested, the user has the option of computing intramolecular terms using the
intramolecular_score
parameter. For the sake of speed, only the interactions between segments is considered. If a segment has not moved, then the contribution of its interaction with the receptor to the intermolecular score is not recalculated. If any two segments have not moved, then the contribution of their interaction to the intramolecular score is not recalculated.
The treatment of flexible molecules will be elaborated further. The first stage of processing is the
Identification of Rigid Segments
. The second stage of processing is the conformation search, at which point the user has the choice of two methods. An
Anchor-First Search
may be selected, in which a molecule conformation is constructed and minimized one segment at a time, starting from an anchor segment. Alternatively, a
Simultaneous Search
will be performed, in which the entire molecule conformation is constructed and minimized in one step.
Identification of Rigid Segments
A flexible molecule is treated as a collection of rigid segments. Each segment contains the largest set of adjacent atoms separated by non-rotatable bonds. Segments are separated by rotatable bonds.
The first step in segmentation is ring identification. All bonds within molecular rings are treated as rigid. This classification scheme is a first-order approximation of molecular flexibilty, since some amount of flexibility can exist in non-aromatic rings. To treat such phenomenon as sugar puckering and chair-boat hexane conformations, the user will need to supply each ring conformation as a separate input molecule.
Additional bonds may be specified as rigid by the user. Please refer to the subsequent section,
Manual specification of non-rotatable bonds
.
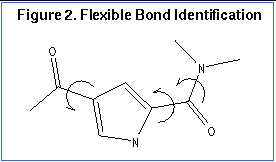 The second step is flexible bond identification, and is illustrated in
Figure 2
for a sample molecule. Each flexible bond is associated with a label defined in an editable file (see
flex.defn on page 110
). The parameter file is identified with the
flex_definition_file
parameter. Each label in the file contains a definition based on the atom types (and chemical environment) of the bonded atoms. Each label is also flagged as minimizable. Typically, bonds with some degree of double bond character are excluded from minimization so that planarity is preserved. Each label is also associated with a set of preferred torsion positions. The location of each flexible bond is used to partition the molecule into rigid segments. A segment is the largest local set of atoms that contains only non-flexible bonds.
The second step is flexible bond identification, and is illustrated in
Figure 2
for a sample molecule. Each flexible bond is associated with a label defined in an editable file (see
flex.defn on page 110
). The parameter file is identified with the
flex_definition_file
parameter. Each label in the file contains a definition based on the atom types (and chemical environment) of the bonded atoms. Each label is also flagged as minimizable. Typically, bonds with some degree of double bond character are excluded from minimization so that planarity is preserved. Each label is also associated with a set of preferred torsion positions. The location of each flexible bond is used to partition the molecule into rigid segments. A segment is the largest local set of atoms that contains only non-flexible bonds.
Manual specification of non-rotatable bonds
The user can specify additional bonds to be non-rotatable, to supplement the ring bonds automatically identified by dock. Such a technique would be used to preserve the conformation of part of the molecule and isolate it from the conformation search. Non-rotatable bonds are identified in the
SYBYL MOL2 format
file containing the molecule. The bonds are designated as members of a STATIC BOND SET named RIGID. Please see
SYBYL MOL2 format on page 99
for an example of such an identification.
Creation of the RIGID set can be done within sybyl. With the molecule of interest loaded into sybyl, select the Build/Edit->Define->Static Set->Bond command. Then select each bond by picking the adjacent atoms. When the "Set Name" dialog comes up, supply the name "RIGID" in capital letters. When the "Comment String" dialog comes up, enter any text you wish. Write out the molecule to file.
Alternatively, the RIGID set can be entered into the MOL2 file by hand. To do this, go to the end of the MOL2 file. If no sets currently exist, then add a SET identifier on a new line. It should contain the text "@<TRIPOS>SET". On a new line add the text "RIGID STATIC BONDS <user> **** Comment". On the next line enter the number of bonds that will be included in the set, followed by the numerical identifier of each bond in the set.
Anchor-First Search
The anchor-first search is an efficient divide-and-conquer algorithm based on the method of Leach and Kuntz [
19
] and the greedy algorithm. It is specified using the
anchor_search
parameter.
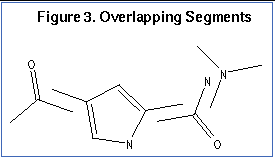 An anchor segment is selected from the rigid segments in an automatic fashion (see
Manual specification of anchor segment
to override this behavior). As illustrated in
Figure 3
for a sample molecule, the molecule is divided into segments that overlap at each rotatable bond. The segment with the largest number of heavy atoms is selected as the anchor. If the
multiple_anchors
parameter is set, then all segments which pass the
anchor_size
cutoff are tried separately as anchors.
An anchor segment is selected from the rigid segments in an automatic fashion (see
Manual specification of anchor segment
to override this behavior). As illustrated in
Figure 3
for a sample molecule, the molecule is divided into segments that overlap at each rotatable bond. The segment with the largest number of heavy atoms is selected as the anchor. If the
multiple_anchors
parameter is set, then all segments which pass the
anchor_size
cutoff are tried separately as anchors.
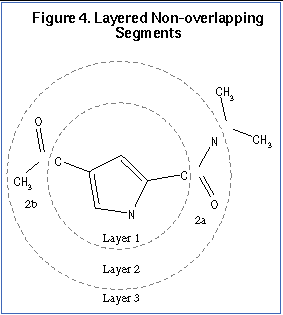 When an anchor has been selected, then the molecule is redivided into non-overlapping segments, which are then arranged concentrically about the anchor segment. This process is illustrated in
Figure 4
for a sample molecule. Segments are reattached to the anchor according to the innermost layer first -- and within a layer -- the largest segment first.
When an anchor has been selected, then the molecule is redivided into non-overlapping segments, which are then arranged concentrically about the anchor segment. This process is illustrated in
Figure 4
for a sample molecule. Segments are reattached to the anchor according to the innermost layer first -- and within a layer -- the largest segment first.
The anchor is processed separately (either oriented, scored, and/or minimized). The anchor position can be optimized prior to the conformation search with the
minimize_anchor
parameter.
The remaining segments are subsequently re-attached during the conformation search. See
Figure 5
for a diagram of the anchor-first docking process. The conformation search corresponds to steps 2 and 3 which form a complete cycle. An extensive analysis of the docking can be performed by setting the
write_partial_structures
parameter which causes all partially-built structures to be written out during the conformation search. Two files will be generated for each cycle of the conformation search. For the first cycle, one file will contain the anchor orientations from step 1 in
Figure 5
and the other file will contain the pruned orientations from step 2. For all subsequent cycles, one file will contain the conformationally-expanded configurations from step 3, and the other file will contain the pruned configurations from step 2. The names of the files will be based on the name given for the ligand output file, but will have a sequence of numbers appended to it as shown in
Table 6
.
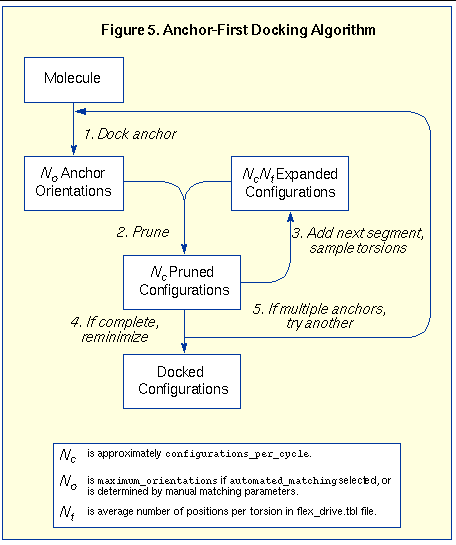
Table 6.
Filename Construction when Writing Partial Structures
|
Files are name-A-LL-S-E.ext, with the components defined as follows:
|
|
name
|
The base file name of the ligand output file.
|
|
A
|
Anchor number (single digit).
|
|
LL
|
Layer number (two digits).
|
|
S
|
Segment number within layer (single digit).
|
|
E
|
Ensemble number within cycle (1=conformation expanded; 2=pruned).
|
|
ext
|
File extension (*.mol2, *.pdb, etc.).
|
If
torsion_drive
has been selected, then the torsion positions of the intervening bond are searched when each segment is reattached. If
torsion_minimize
has been selected, then the intervening torsion may be relaxed. Minimization of the bond is performed in isolation, or in concert with inner torsions if the
reminimize_layer_number
parameter is set to a non-zero value. Relaxing multiple layers helps prevent the search from getting stuck in dead-ends. Although computationally expensive, the position of the anchor may be simultaneously optimized during the conformation search with the
reminimize_anchor
parameter. When all segments have been added, the entire molecule may be relaxed if the
reminimize_ligand
parameter is set.
Docking with the Anchor-First procedure
The process of docking a molecule using the anchor-first strategy is shown in
Figure 5
. The amount of searching is under full user control. The process begins with docking the anchor. This step is controlled with the
orient_ligand
parameters (please refer to
Orientation Search on page 28
) and results in No anchor positions. The conformation search begins by pruning these orientations according to rank and position (see
Pruning the conformation search tree
below) to produce Nc positions. Each subsequent cycle of the conformation search involves expanding the ensemble of partially built binding positions by adding a new segment and performing a torsion search on the newly formed bond and then contracting the ensemble with pruning. The torsion search on each newly formed bond results in an expansion of the set of Nc partial configurations to NcNt configurations. The size of Nt is based on the number of increments used for the current bond and can be modified by altering the entries in the
flex_drive.tbl
file. The expanded set of binding positions is then pruned back to Nc configurations. The conformation search continues expanding and pruning the set of partial binding position until each binding position represents a complete molecule.
This search technique is particularly useful for docking, but it also may be used for conformation analysis and stand-alone minimization.
Pruning the conformation search tree
During each cycle of the conformation search, the expanded set of partial configurations is pruned based on the setting of
configurations_per_cycle
. The pruning attempts to retain the best, most diverse configurations using a top-first pruning method which proceeds as follows. The configurations are ranked according to score. The top-ranked configuration is set aside and used as a reference configuration for the first round of pruning. All remaining configurations are considered candidates for removal. A weighted root-mean-squared distance (wRMSD) between each candidate and the reference configuration is computed according to
Equation 1
.
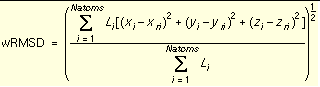 Equation 1
Equation 1
where Li is the layer to which atom i is assigned. The RMSD is weighted in this fashion to make it more sensitive to the position of the outer segments. The outer segments are more important because they have a greater influence over the position of subsequently added segments.
Each candidate is then evaluated for removal based on its rank and wRMSD using the inequality shown in
Equation 2
. If the factor is greater than
configurations_per_cycle
, the candidate is removed. Based on this factor, a configuration with rank 2 and 0.2 Angstroms wRMSD is comparable to a configuration with rank 20 and 2.0 Angstroms wRMSD. The next best scoring configuration which survives the first pass of removal is then set aside and used as a reference configuration for the second round of pruning, and so on.
 Equation 2
Equation 2
This pruning method attempts to balance the twin goals of recovering the best scoring and the most different binding configurations without introducing additional user parameters. The pruning method replaces the hierarchical clustering method used in the initial release of dock version 4.0, because it is faster (N logN versus N2) and biases its search time towards molecules which sample a more diverse set of binding modes. As the value of
configurations_per_cycle
is increased, the anchor-first method approaches an exhaustive search.
Time requirements
The time demand grows linearly with
configurations_per_cycle
, the number of flexible bonds and the number of torsion positions per bond, as well as the number of anchor segments explored for a given molecule. Using the notation in
Figure 5
, the time demand can be expressed as
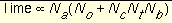 Equation 3
Equation 3
where the additional terms are:
|
Na
|
is the number of anchor segments tried per molecule.
|
|
Nb
|
is the number of rotatable bonds per molecule.
|
Manual specification of anchor segment
The user can override the automatic anchor selection performed by dock by specifying a STATIC ATOM SET named ANCHOR in the molecule input file. For an example, please see
SYBYL MOL2 format on page 99
. See the previous discussion of
Manual specification of non-rotatable bonds
for related instructions. It must be pointed out that the user can include as many atoms as desired in the ANCHOR set, but only the first atom will be used. The anchor segment which includes the ANCHOR atom will then be used as the segment anchor. In order to make a larger anchor than would be produced using the automatic segmentation based on the location of rotatable bonds, the user will need to manually specify the necessary bonds as non-rotatable (with a RIGID BOND set).
Simultaneous Search
If an anchor-first search is not selected, then a simultaneous search is performed by default. All torsions are searched and/or minimized in concert. The conformation search is performed prior to the orientation search, so each conformation is docked independently. The simultaneous search technique is useful for conformation analysis and for constructing a chemical screen database (see
Chemical Screen on page 44
).
Constraining the search
During a simultaneous search, dock performs an exhaustive or a random search, depending on the flexibility of the molecule and the value of
conformation_cutoff_factor
. The cutoff on the number of conformations generated for a molecule is calculated by
Equation 4
.
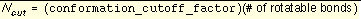 Equation 4
Equation 4
If the total conformations for a molecule is below Ncut an exhaustive search of all conformations is performed. Otherwise, a random search of Ncut conformations is performed, in which torsion positions are selected randomly from the allowed positions. This definition of Ncut was used so that the time demand grows linearly with the number of rotatable bonds, which is a fair compromise between the exponential growth of the total possible conformations and a fixed cutoff applied uniformly to all molecules.
Caveat on backtracking procedure
A backtracking procedure is used during the generation of each conformation. If an internal clash is detected for the torsion position of a rotatable bond within a partially-built structure, then another torsion value is attempted. If no torsion values will work, then the procedure backtracks to the preceding rotatable bond and assigns a new torsion for it. This procedure works fine when internal clashes can be resolved easily, but in the worst case, for a molecule with a huge number of rotatable bonds and a clash that cannot be resolved at the last rotatable bond, the procedure will be confounded and tend to consume cpu time. For this reason, when processing a database of molecules, be sure to use a reasonable value of
flexible_bond_maximum
to discard overly flexible molecules.
Torsion Minimization
The torsion angles of rotatable bonds may be included in the
Score Optimization
. This provides for a much more efficient conformation search since fewer torsion positions need to be sampled. Each torsion flagged for movement is assigned a simplex vertex along with the six rigid body degrees of freedom. Only non-bonded interatomic terms are included in the scoring evaluation; no explicit torsion terms are included. Therefore, only torsions flagged as minimizable in the
flex.defn
file are included (e.g. double bonds are excluded by default). When an
Anchor-First Search
is performed with segments from multiple layers being minimized, then inner torsions are assigned smaller initial torsion step values since perturbations in these torsions have a greater impact on conformation.
© UC Regents 1998
Top / Up / Previous / Next