 ,
interleukin-1, and interleukin-3 (Marx, 1987; Angel and Karin, 1991; Smith
et al., 1993).
,
interleukin-1, and interleukin-3 (Marx, 1987; Angel and Karin, 1991; Smith
et al., 1993).
Oncoproteins c-Jun and c-Fos
Francis H. C. Crick, What Mad Pursuit (1988)
Our knowledge of molecular biology has progressed far since Watson and Crick first proposed their structure of DNA (Watson and Crick, 1953b). At that time they correctly predicted the mode of DNA self-replication and that it was the sequences of bases which carried the genetic information (Watson and Crick, 1953a; 1953c). We now possess a much more comprehensive knowledge of how the cell replicates its DNA and how it decodes the information contained in the gene into protein, or to quote James Watson's `central dogma': DNA->RNA->protein (Watson, 1970). However, one enigma that Watson and Crick did not initially address and that has been slow to yield to modern scientific study is that of differential gene expression.
Almost every cell in the human body carries the same complement of DNA, i.e., they share the same genotype, yet human cells display a very large range of differing phenotypes. For example, a macrophage from one individual looks and behaves differently to a hepatocyte from the same individual despite the fact that the two cells share the exact same set of genes. These differences arising from a background of genetic identity are possible because cells have the ability to express a subset of their genome. Furthermore cells are able to express different portions of their genome at different times depending upon their needs. In other words, organisms have the ability to regulate gene expression. This is a critical property of all living systems as it allows organisms to adapt to changes in their environment.
 has a
fundamental decision to make upon infecting
an Escherichia coli cell; it must either lyse or lysogenise the host.
This seems like a rather insurmountable task for something without a cognitive
ability. However, phage
successfully makes this decision every
time it infects a new cell. The decision is made by the presence or absence of
certain proteins within the bacterial cell, which in turn results in either the
phage genes for lysis being
turned on and those for lysogeny
being turned off, or vice versa (for a full description see Ptashne
(1986)).
has a
fundamental decision to make upon infecting
an Escherichia coli cell; it must either lyse or lysogenise the host.
This seems like a rather insurmountable task for something without a cognitive
ability. However, phage
successfully makes this decision every
time it infects a new cell. The decision is made by the presence or absence of
certain proteins within the bacterial cell, which in turn results in either the
phage genes for lysis being
turned on and those for lysogeny
being turned off, or vice versa (for a full description see Ptashne
(1986)).Gene regulation also plays an important role in viruses which infect eukaryotic cells. The human immuno-deficiency virus is a good example of an organism which produces undesirable gene activity. Treatment of such a disease is particularly difficult because the proviral genome is integrated into the host DNA. The onset of clinical symptoms can occur years after the initial infection, during which time the virus lays dormant. This period of dormancy, during which the level of virus particles in the circulation is so low as to be undetectable, indicates that the proviral genome is under tight transcriptional regulation. Although death is usually the result of a secondary infection, it is immune system failure which is the underlying cause. This immune system failure is associated with increased viral activity which is a result of a change in the regulation of the proviral genome. Therefore factors which can inhibit HIV gene expression form potential therapies.
Gene regulation plays a vital role throughout the lifetime of all organisms. The embryos of higher organisms switch on and off whole arrays of genes during the course of their development, with individual cells making decisions which will ultimately determine their role in the mature organism. However, aberrant gene regulation can lead to disease states. One good example of such a disease is cancer, in which the affected cells continually multiply where their normal counterparts do not. This alteration in growth pattern can be traced to the activity of a group of genes called oncogenes, i.e., genes which cause cancer. Some oncogenes are activated by alterations to their structure. These alterations can take many forms, including changes to the protein sequences encoded, changes to portions of the mRNA transcripts outside of the polypeptide coding regions, and changes to the promoter, enhancer and other regions which control gene expression. The proteins encoded by oncogenes (oncoproteins) are diverse in both structure and function, and include growth factors, cell surface receptors, protein kinases, and DNA-binding proteins. Despite this diversity of activities, oncoproteins share the common function of being involved with the signal transduction pathways that are responsible for converting extra- and intracellular signals into changes in cell growth. These pathways all culminate in the cell nucleus with the modulation of the activities of proteins which bind to DNA and alter gene transcription, the ultimate outcome being that certain genes are continually switched on (or off) and hence the cells divide uncontrollably. Thus, these transcription factors represent one key connection between growth signals and the resulting cell division. Cancer presents us with a good working model of abnormal gene regulation. By comparing the cancerous state with the normal state, it is possible to isolate the factors, such as transcription factors, which are involved with the regulation of cell growth. Thus, the study of transcriptional regulation can be very useful, not only for gaining a greater understanding of the life process, but also from the clinical perspective of developing treatments for many life-threatening diseases. Following Francis Crick's assertions given in Section 1.1.1, these studies must be made at the molecular level if we are to be confident in our understanding of how gene regulation is achieved.
Transcription of a eukaryotic gene encoding a protein is initiated with binding of various factors to the promoter region of the gene, located 5' to the coding region; RNA polymerase II cannot by itself recognise a promoter and initiate transcription. Once these factors are bound, RNA polymerase II can bind to the promoter, and the stable complex thus formed cannot be easily displaced from the DNA. Transcription is then initiated with the hydrolysis of ATP to ADP. This hydrolysis is distinct from that required for the incorporation of ATP into the nascent RNA chain. Transcription then proceeds until termination occurs beyond the coding region. The exact mechanisms involved in the termination reaction are poorly understood. However, most mRNAs are cleaved at their 3' end near the conserved sequence AAUAAA which acts as a signal for the cleavage and subsequent polyadenylation of the 3' tail. The nascent pre-mRNA also undergoes 5' capping and splicing before it is translocated to the ribosomes for translation.
Another location for gene regulation is enhancers sequences. These DNA sequences can be located up to several thousand base pairs away from a gene and still have the ability to augment its transcription by over a factor of 100. How this is achieved over such long distances is still unclear. Enhancer sequences are often long (hundreds of base pairs) and are usually comprised of repeat sequences. An enhancer can lie in either orientation with respect to a gene and it can be either 5' or 3' of the promoter. Interestingly, enhancers can even lie within the genes they regulate. Enhancers are cis-acting elements, lying on the same DNA molecule as the gene they affect i.e., they do not work between chromosomes. One enhancer can alter the levels of expression of more than one gene, but it will preferentially act on the closest one. Enhancers are also tissue specific and thus are likely to be important in differential expression during development.
Enhancers exert their activities by binding proteins which must in some way make the DNA in the vicinity of a nearby promoter more accessible to the binding of RNA polymerase II and/or associated factors. A possible model of gene activation developed by Ptashne and Gann (1990) is shown in Fig. 1.1. Enhancer binding proteins belong to the group of proteins known as gene transcription factors. They are able to act like molecular switches, turning the expression of genes on or off in response to the needs of the cell and in turn to the needs of whole organisms. Many of these transcription factors have been identified as being the products of oncogenes. One such oncoprotein is called c-Jun and it forms the basis of the work described here.
Investigation of AP-1 quickly revealed that it is composed of several distinct polypeptides (Angel et al., 1987; Lee et al., 1987b). These polypeptides have now been identified as the products of the jun and fos gene families (Curran and Franza, 1988; Angel and Karin, 1991). Cotransfection of a plasmid encoding either c-jun or v-jun, with a TRE-containing indicator plasmid, into undifferentiated F9 embryonal carcinoma stem cells (which show negligible AP-1 activity and undetectable c-jun and c-fos transcription) results in specific activation of the reporter gene on the indicator plasmid (Angel et al., 1988). By comparison, cotransfection of c-fos alone results in only marginal transactivation. However, cotransfection of c-jun and c-fos simultaneously produces more potent transactivation than can be achieved with c-jun alone (Chiu et al., 1988; Sassone-Corsi et al., 1988). This behaviour is a consequence of the need for these proteins to dimerise before they can bind to DNA. c-Jun is capable of homodimerising, and hence is active in the cotransfection experiment. Conversely, c-Fos is incapable of forming homodimers under physiological conditions and thus cannot bind to DNA on its own (Kouzarides and Ziff, 1988; Nakabeppu et al., 1988). The increased stimulation of AP-1 activity seen when both c-jun and c-fos are present is a consequence of the formation of c-Jun:c-Fos heterodimers which are more efficient at binding DNA than c-Jun homodimers (Angel and Karin, 1991).
AP-1 activity is displayed not only by c-Jun homodimers and c-Jun:c-Fos heterodimers, but also by a whole range of different dimers formed by members of the Jun and Fos protein families. Thus the term AP-1 is now used to describe the binding activity of these protein complexes to the TRE (Abate and Curran, 1990). The connection of c-jun and c-fos with AP-1 has lead to these two gene families and their products becoming the subject of intensive scientific study.
The expression of both the normal cellular genes c-jun and c-fos
can be induced in the presence of protein synthesis inhibitors (i.e.,
without the need for de novo protein synthesis). This has led to them
being classified as immediate early genes. They are rapidly induced in response
to a broad range of external growth signals and they are responsible for
converting these short-term cell growth and differentiation signals,
originating from outside the cell, into long-term, programmed responses by the
cell (Vogt et al., 1990). Indeed, c-fos gene activity is believed
to be so crucial in this role that it has been dubbed the "master switch"
(Marx, 1987). It makes the earliest known nuclear response to numerous growth
factors including platelet derived growth factor, epidermal growth factor,
fibroblast growth factor, and nerve growth factor as well as tumour-promoting
phorbol esters, colony-stimulating factor-1, tumour necrosis factor ,
interleukin-1, and interleukin-3 (Marx, 1987; Angel and Karin, 1991; Smith
et al., 1993).
Since the discovery of c-jun and c-fos, several related genes have been isolated which reveal that these genes are members of two gene families. The fos gene family is currently known to comprise c-fos, fosB, fra1, fra2 and dFRA while the jun family is made up of c-jun, junB, junD and dJRA. Just like c-fos, all the members of the fos family form part of the immediate early response. This is not true of the jun family, with only c-jun and jun B being responsive to mitogens. The products of these genes display different functions with some acting as transcriptional activators, while others act as transcriptional repressors.
c-Jun and c-fos play a vital role in the coupling of external signals for growth and differentiation to changes in gene transcription. A general model of stimulus-response coupling, proposed by Curran and Morgan (1987), is shown in Fig. 1.2. In this model, extracellular stimuli, such as the binding of growth factors to cell-surface receptors, results in an alteration in the levels or activity of secondary messenger molecules. These secondary messengers elicit immediate short-term responses as well as stimulating the expression of genes encoding adaptive regulators, such as c-jun and c-fos, whose products then modulate the expression of genes which produce long-term responses. These adaptive regulators would also control the expression of components in the secondary messenger systems to alter the cells response to subsequent stimulation. Thus c-jun and c-fos, along with other immediate early genes, represent the linchpin between growth signals and the ensuing gene expression required to produce cell growth and differentiation.
c-Jun and c-Fos are comprised of several functional domains which are depicted in Fig. 1.3. The best characterised domains are those that mediate dimerisation and DNA binding, found near the middle of c-Fos and within the C-terminal portion of c-Jun. These domains are central to the activity of these proteins as transcriptional activators and are described in more detail later in this chapter. c-Jun and c-Fos also contain domains that can regulate both their DNA-binding activity and their ability to stimulate transcription.
c-Jun contains two regulatory domains known as A1 and A2, which lie N-terminal to the dimerisation and DNA-binding domains. A1 contains two further interdependent activation regions which share homology with identically spaced sequences in c-Fos, known as Homology Boxes 1 and 2 (see Fig. 1.3) (Hurst, 1994). The activity of these domains is regulated by kinase-specific phosphorylation of serine and threonine residues. This regulation is positive if phosphorylation occurs in the A1 domain at either of two sites which lie within and just adjacent to the HOB1 (Fig 1.3). Conversely, negative regulation of c-Jun occurs when sites N-terminal to the DNA-binding domain are phosphorylated, which results in a decrease in DNA-binding activity. DNA-binding can also be inhibited by the oxidation of a conserved cysteine residue located within the DNA-binding domains of both c-Jun and c-Fos (Abate et al., 1990). This oxidation results in the formation of sulfhydryl oxidation products which interfere with the cysteine-sidechain/DNA interactions and does not involve the formation of disulfide bonds.
c-Fos contains a transrepression domain within its C-terminal 27 amino acids.
Phosphorylation of sites within this domain permits c-Fos to transrepress
expression of the c-fos gene in a manner which is independent of the
DNA-binding domain. v-Fos lacks these phosphorylation sites and is thus unable
to transrepress v-fos expression. In addition to phosphorylation,
repression can be mediated by alternative splicing products of the fully
functional forms of these transcriptional activators. For example, one form of
FosB, known as 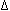 FosB, lacks the
C-terminal 101 amino acids which is
postulated to contain an activation domain (Foulkes and Sassone-Corsi, 1992).
However, it still retains the protein dimerisation and DNA-binding domains
which permit it to form inactive DNA-binding heterodimers with c-Jun. Thus the
fosB gene can produce both an activator and a repressor from the one
coding sequence.
FosB, lacks the
C-terminal 101 amino acids which is
postulated to contain an activation domain (Foulkes and Sassone-Corsi, 1992).
However, it still retains the protein dimerisation and DNA-binding domains
which permit it to form inactive DNA-binding heterodimers with c-Jun. Thus the
fosB gene can produce both an activator and a repressor from the one
coding sequence.
The transformationally active form of c-Jun, v-Jun, contains several alterations and point mutations at locations which correspond to the regulatory domains of c-Jun. One substantial alteration is the removal of a fragment, called d, from the A1 domain. d is believed to bind a cell-type specific repressor. Interestingly, d is located immediately adjacent to the two phosphorylation sites involved with the activation activity of the A1 domain. Thus binding of a repressor to the d sequence in c-Jun may act to block the activity of protein kinases on these sites. v-Jun also has two point mutations, the first is a mutation at Ser243 to a Phe, making its DNA binding activity immune to down regulation by phosphorylation (Smith et al., 1993). The second, a mutation at Cys269 to a Ser, renders v-Jun immune to down regulation by the redox regulation mechanism mentioned earlier (Abate et al., 1990). Thus the net effect of these alterations within v-Jun are to render it largely immune to the effects of down regulation, resulting in v-Jun being continually active.
The significance of the heptad repeat is not immediately obvious until the
sequence is modelled as an -helix. Inspection of these sequences in
the form of a helical wheel diagram (see Fig.
1.5) shows that the leucine
residues align along one face of the hypothetical helix. Even though these
protein sequences are somewhat dissimilar, they all show notable amphipathy
when modelled as helices, one side of the helix being predominantly composed of
hydrophobic residues (particularly leucine), while the other comprises a
mixture of both acidic and basic residues, as well as uncharged polar residues.
This arrangement is characteristic of proteins which form long stable
-helices and can lead to
intertwining of the helices to form
coiled-coils, as seen in the keratins, lamins, and paramyosin (Crick, 1952;
Cohen and Holmes, 1963; Parry et al., 1977; McKeon et al., 1986).
Helix formation is stabilised by the amphipathic arrangement of hydrophobic
residues and the formation of salt bridges between sidechains of opposite
charge (Schulz and Schirmer, 1979; Chothia, 1984; Sundaralingam et al.,
1987). Landschulz (1988) observed that C/EBP is not only amphipathic, but that
it is also rich in oppositely charged residues that are juxtaposed in a manner
suitable for intrahelical ion pairing and thus concluded that it was highly
probable that this sequence exists as a stable -helix in aqueous
solution.
Bearing these facts in mind, Landschulz et al. (1988) proposed that dimerisation of these proteins was mediated by leucyl sidechains, protruding from one monomer, interdigitating with the corresponding leucyl sidechains of another monomer in a fashion similar to the interlocking of teeth in a zipper (see Fig. 1.6) and thus coined the term `leucine zipper'. Furthermore, they suggested that the orientation of the helices with respect to one another would be antiparallel. Computer modelling of the C/EBP leucine repeat motif showed the leucine sidechains to be disposed at an angle pointing towards the amino terminus. Thus an antiparallel orientation would be more amenable to the interdigitation of these sidechains. The antiparallel arrangement would also allow the helix dipoles to attract, rather than repel. However, the possibility of a parallel arrangement was not excluded.
The parallel orientation of the GCN4 leucine zipper combined with the
observation of a 4-3 repeat of hydrophobic residues within the motif
(i.e., the leucine residues lie four residues before and three residues
after another hydrophobic amino acid) strongly suggested that dimerisation was
achieved via the formation of a coiled-coil of -helices (O'Shea et
al., 1989a).
-helices in solution (see Fig. 1.7). This symmetry, combined
with the fact that the helices are parallel (see Section
1.3.2), gave further
support to the hypothesis that leucine zippers form coiled-coils, as it is
impossible for two parallel leucine zippers to have their leucyl sidechains
interdigitate and still form a symmetric dimer (see
Fig. 1.8). However, the
complete symmetry observed in the NMR spectra precluded calculation of dimer
structures using conventional methods.
The coiled-coil hypothesis was confirmed by X-ray scattering of two crystal
forms of a 33-residue peptide corresponding to the leucine zipper of GCN4
(Rasmussen et al., 1991). Comparison of the GCN4 crystal's diffraction
patterns with the pattern obtained from the coiled-coil protein
-keratin revealed strong
similarities. Both GCN4 crystals show strong
meridional reflections at
5.15-5.2 Å rather than at 5.4 Å resolution which would be
expected for straight -helices.
They also share strong equatorial
reflections on the equator at 10 Å. These reflections are also in
agreement with Crick's (1953a; 1953b) calculations of reflections from
hypothetical coiled-coils.
The monoclinic form of these GCN4 leucine zipper crystals diffracted X-rays to
1.8 Å resolution and the data thus obtained was used to obtain a
high resolution structure of the dimer(O'Shea et al., 1991). This
structure (see Fig. 1.9 and 1.10) revealed that the two parallel
-helices formed a classical
coiled-coil which was consistent with
Crick's `knobs-into-holes' model of helix packing (1953b). In the crystal, each
-helix wraps around the other to
produce a left-handed superhelix. The
crossing angle of the two helices is ~18 (very close to Crick's (1953b)
suggested value of 20). The
residues of the 4-3 hydrophobic repeat lie
against one another forming a structure which resembles a ladder, the sides
being formed by the helical backbones and the hydrophobic sidechains forming
the rungs.
(very close to Crick's (1953b)
suggested value of 20). The
residues of the 4-3 hydrophobic repeat lie
against one another forming a structure which resembles a ladder, the sides
being formed by the helical backbones and the hydrophobic sidechains forming
the rungs.
Thus, although the original leucine zipper hypothesis (Landschulz et
al., 1988) correctly predicted that the leucine zipper motif was a protein
dimerisation element, that the leucine residues were vital to this function,
and that it formed an extended -helix, it was incorrect in predicting
the mode of the helical association as being antiparallel and that the leucine
residues interdigitated like the teeth of a zipper. Ironically, the leucine
zipper model was based upon the coiled-coil paradigm. It was the almost
exclusive use of leucine, which is unusual for coiled-coils, that lead
Landschulz et al. (1988) to formulate their incorrect zipper
hypothesis.
One possible explanation for the conservation of leucine is that it was the heptad repeat that was used as the criterion for identifying leucine zipper proteins (O'Shea et al., 1991). Indeed there are a number of proteins which contain two non-leucine residues at the leucine position (e.g., CPC-1, dC/EBP, CRP-1). However, this still cannot account for the very large number of proteins which do show the very strong conservation of leucine. The consistent use of a leucine repeat may facilitate the formation of heterodimers. In this role leucine would act as adaptor between leucine zipper monomers, providing a common interface and thus a basal level of affinity, with residues at other positions in the heptad determining the overall level of affinity (O'Shea et al., 1991). The use of leucine on only one face of the helix may also favour the formation of parallel coiled-coils which is required to bring the basic domains into juxtaposition for DNA binding (Alber, 1992).
However, a more fundamental explanation for the use of leucine lies in the fact that the coiled-coil formed by leucine zippers is incredibly short (approximately one quarter to one third of a superhelical turn) by comparison with fibrous proteins such as keratin which extend over many superhelical turns. In these fibrous proteins, leucine makes up only one-quarter to one-half of the residues found at the equivalent position within the heptad repeat (Parry, 1982; Conway and Parry, 1990). The short lengths of leucine zipper dimer interface would require the use of especially stabilising residues. The crystal structure of the GCN4 leucine zipper (O'Shea et al., 1991) shows that leucine does act as a stabilising agent as it is able to fill more space between the helices, it packs well with adjacent residues, and makes closer contacts with adjacent layers than other hydrophobic sidechains do. Thus, leucine is most probably the ideal amino acid for stabilising short coiled-coils.
One not so obvious reason is the effect of other types of residues on the oligomerisation state of coiled-coils. Francis Crick (1953b), in his postulation of the coiled-coil, hypothesised that the knobs-into-holes paradigm could easily be extended to the formation of three-stranded coiled-coils. Modification of the GCN4 leucine zipper which uniformly replaces all of the hydrophobic interfacial residues with isoleucine results in a molecule which forms a three-stranded coiled-coil (Harbury et al., 1993; 1994). Various other combinations can also produce four-stranded coiled-coils. Thus, leucine in combination with the other residues of the 4-3 hydrophobic repeat may be critical in maintaining the dimer state over other higher-order oligomers. This is important because dimerisation is critical to the DNA binding activity of transcriptional activator proteins which contain leucine zippers.
-helices within the major groove
of DNA to make specific contacts with
the bases. These contacts include hydrogen bonds, either direct or water
mediated, and non-polar van der Waals interactions. Other contacts outside the
major groove, with atoms of the sugar-phosphate backbone, are critical to the
positioning of the domains within the major groove (Harrison, 1991).
Prior to the discovery of the leucine zipper proteins, two main groups of
DNA-binding motifs had been elucidated: the helix-turn-helix and the Zn-finger.
The helix-turn-helix (HTH) motif is characterised by two helices that are
separated by a 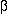 -turn. The HTH
motif was first identified and
characterised in prokaryotic activator and repressor proteins. It shows
considerable sequence variability, but maintains a highly conserved geometry
with the recognition helix directly contacting bases within the major groove of
the target DNA, while the other -helix, positioned at nearly right
angles to the recognition helix, lies across the major groove and makes some
non-specific contacts (Struhl, 1989). Eukaryotic examples of this group include
the homeodomain proteins Antp (Otting et al., 1990) and Engrailed
(Kissinger et al., 1990).
-turn. The HTH
motif was first identified and
characterised in prokaryotic activator and repressor proteins. It shows
considerable sequence variability, but maintains a highly conserved geometry
with the recognition helix directly contacting bases within the major groove of
the target DNA, while the other -helix, positioned at nearly right
angles to the recognition helix, lies across the major groove and makes some
non-specific contacts (Struhl, 1989). Eukaryotic examples of this group include
the homeodomain proteins Antp (Otting et al., 1990) and Engrailed
(Kissinger et al., 1990).
Two different Zn-finger motifs have been found to date. The first (class I) is
an approximately 30-residue module which coordinates one Zn ion between two
cysteines and two histidines. The second (class II) is an approximately
70-residue module which binds two Zn ions, each coordinated by four cysteines
each. The class I Zn-fingers are composed of a short -helix packed
against a -hairpin. They bind to
DNA as a repeating structure with
three or more fingers in direct succession. The main contacts with DNA are made
by the short -helices lying in
the major groove, contacting three base
pairs each. The repeating Zn-finger chain wraps around the DNA with a three
base pair gap between each Zn-finger (Pavletich and Pabo, 1991). The class II
Zn-fingers form a loop-helix-loop-helix structure with the two helices lying
almost perpendicular to one another. One helix lies within the major groove
when bound to DNA and sidechains from the second helix interact with the DNA
bases (Luisi et al., 1991). The second helix and the loops make
non-specific positioning contacts with the sugar-phosphate backbone in a
fashion similar to that of the HTH motif.
-helices and the
bifurcating arms of the Y being formed by the basic domains which, like the
leucine zipper motif, form extended -helices. These basic domain
helices then lie within the major grooves at the centre of the dyad-symmetric
recognition site.
The induced helical fork also predicted that the helical conformation of the
basic domain is induced in the presence of DNA and that the
N--AA--(C/S) quartet consensus sequence was critical for DNA
binding. The scissors-grip model suggested that the basic domain contained an
N-cap at the N-terminal Asn of the consensus sequence. An N-cap is formed by
the sidechain oxygen of Asn hydrogen bonding to the peptide nitrogen two or
three residues into an -helix,
thus forming an N-terminal cap to the
helix (Presta and Rose, 1988; Richardson and Richardson, 1988). This N-cap in
the basic domain would allow the domain to form a 75 bend between two
-helices, which in turn would
permit residues N-terminal of the
consensus sequence to continue tracking around the major groove of the DNA
binding site. Thus, the structure formed is similar to the grip of a wrestler
grabbing the torso of his opponent with the 75 bend corresponding to his
knees, hence the term `scissors-grip' (Vinson et al., 1989). The need
for a bend was deemed important as mutations of residues N-terminal to the
consensus sequence block DNA binding (Gentz et al., 1989; Landschulz
et al., 1989; Neuberg et al., 1989; Turner and Tjian, 1989). This
model required that the formation of the second helix N-terminal to the N-cap
and/or the N-cap itself be formed only upon DNA binding, to permit the protein
to dynamically engage and disengage.
Several lines of experimental evidence have given support to these models. DNA methylation protection and hydroxyl radical cleavage footprinting show that C/EBP does bind its cognate DNA site in a symmetrical manner (Vinson et al., 1989), as predicted in both models. Mutation of the Asn residue, hypothesised to form an N-cap in the scissors-grip model, results in a marked loss of DNA-binding activity in the c-Fos bZIP domain (Neuberg et al., 1991). Construction of a minimal bZIP domain based upon the N--AA--(C/S) quartet consensus sequence and idealised coiled-coils results in a peptide which is able to bind DNA in a sequence-specific manner (O'Neil et al., 1990).
Circular dichroism (CD) studies on both native GCN4 and the bZIP domains of
GCN4 and C/EBP (Patel et al., 1990; Weiss 1990b; Weiss et al.,
1990; O'Neil et al., 1991) show that the basic domain is largely
unfolded in the absence of DNA and that its folded, -helical
conformation is stabilised by binding to DNA. Analysis of thermal unfolding and
refolding indicates that there are three separate states for the GCN4 bZIP
domain: (I) fully unfolded monomers, (II) partially folded dimer state in which
the coiled-coil of the leucine zipper has formed, and (III) fully folded state
which occurs only upon sequence-specific DNA binding (Weiss, 1990b). NMR
studies of both the isolated basic domain and the entire bZIP domain of GCN4 in
the absence of DNA shows that the basic domain forms a mobile flexible segment
that folds into a loose helix (Saudek et al., 1990; 1991a). Thus many
aspects of both the scissors-grip and the induced helical fork models appear to
be correct.
-helices
and straight B-DNA, indicating that either the protein, and/or the DNA must be
bent to accommodate the number of contacts observed (Kerppola and Curran,
1991a). Quantitative application of circular permutation analysis, which
measures DNA distortions based upon the electrophoretic mobilities of DNA
fragments, has shown that c-Jun:c-Fos heterodimers and c-Jun homodimers bend
DNA in opposite orientations (Kerppola and Curran, 1991a; 1991b). This result
suggests that the c-Jun and c-Fos basic domains adopt different structures upon
DNA binding. This led to the formulation of the flexible hinge model (Kerppola
and Curran, 1991a) in which c-Jun binds to DNA in a fashion similar to the
scissors-grip model with an N-cap producing a bend between two helices in the
basic domain. However, the angle between the two helices differs from that
suggested by Vinson et al. (1989) due to the fact that the DNA is bent
towards the dimer interface. In contrast c-Fos bends DNA away from the dimer
interface. The flexible hinge model suggests that this is achieved by the major
groove adopting an extended linear shape over the length of the recognition
site and hence c-Fos does not need to bend within its basic domain. Molecular
structure determination of c-Jun and c-Fos bound to DNA is yet to test the
validity of this model.
and a translation of
3.4 Å of its two half-sites with respect to one another by
comparison with the AP-1 site. The binding of GCN4-bZIP to these two different
sites is facilitated by DNA flexing which can accommodate the protein structure
(König and Richmond, 1993).
In the GCN4-bZIP:AP-1 complex (Ellenberger et al., 1992), 52 out of 58
residues in each monomer form a continuous -helix with no pronounced
sharp bends or kinks (see Fig. 1.12). The
helices pack tightly against one
another in a coiled-coil structure at the C-terminus. The last intermonomer
contact is made between the methionine residues which are three residues
N-terminal to the first zipper-motif leucines. The N-terminal DNA-binding
domains then gently splay to either side of the DNA, traversing the major
groove of each half-site. Various contacts are made between positively charged
and polar residues to unesterified oxygens of the phosphodiester backbone via
hydrogen bonds. The N-terminus continues on as a straight helix past the point
of DNA contact, giving the protein the appearance of a pair of
-helical tweezers.
The binding of the two half-sites is asymmetric, with an arginine of one
monomer contacting the central G (see Fig.
1.11) while the corresponding
arginine of the other monomer hydrogen bonds to unesterified phosphate oxygens.
This causes the GCN4-bZIP dimer to be slightly displaced with respect to the
pseudodyad of the AP-1 site and one monomer is drawn closer to the DNA than the
other. This has the effect of tilting the axis of the coiled-coil by 3 away
from perpendicular with the DNA axis. However, this asymmetry is not propagated
down the helices; instead, local corrections within each monomer result in the
other protein-DNA contacts being common to both half-sites. This indicates that
the helical fork at the junction of the basic domain and the leucine zipper is
somewhat flexible. The AP-1 binding site adopts a regular, straight B-form,
with no systematic variation in phosphodiester backbone or base pair
geometry.
The GCN4-bZIP:ATF/CREB complex (König and Richmond, 1993) shows an overall
structure which is not too dissimilar to the GCN4-bZIP:AP-1 complex, with 49
out of 62 residues adopting an -helical conformation in each monomer
(see Fig. 1.13). These two -helical monomers form a Y-shaped
molecule,
free of any sharp bends or kinks with the arms of the Y splayed by 25 as
opposed to the 20 angle seen in
GCN4-bZIP:AP-1. The C-terminal portion of
the GCN4-bZIP dimer forms a quarter turn of left-handed parallel coiled-coil
with the last intermonomer hydrophobic contact being made by the same
methionine sidechains mentioned previously. Unlike the GCN4-bZIP:AP-1 complex,
the coiled-coil lies exactly perpendicular to the DNA axis.
Despite the anticipation of major differences between the AP-1 and ATF/CREB
complexes within the DNA/protein contact region, the two structures appear
highly similar. The complete dyad symmetry of the ATF/CREB site allows the
basic domains to bind in a symmetric fashion. However, the DNA is distorted
away from the B-form by a symmetric 20 bend at the centre of the site
towards the coiled-coil to accommodate the extra G.C pair, resulting in a
slight deepening and narrowing of the major groove. The DNA is also marginally
unwound from 10.5 to 11.0 base-pairs per turn. In effect the ATF/CREB site is
altered to mimic the spatial relationships of the bases on one strand seen in
the AP-1 site. The interactions between GCN4-bZIP and the other strand are then
displaced by one base, although, the same specific interactions are maintained.
Thus, the
DNA structure flexes to accommodate the more rigid protein structure.
These structures most resemble the induced helical fork model (O'Neil et al., 1990), differing only slightly in the details of some of the protein-DNA contacts. In contrast, the scissors-grip model (Vinson et al., 1989) does not compare favourably with the X-ray structures. The anticipated bending of the basic-domain helices arising from an N-cap is not evident. In both structures, the invariant asparagine hydrogen bonds exclusively to DNA bases (König and Richmond, 1993). The scissors-grip model, although now apparently unlikely, may still be viable as it can explain the much greater bending of DNA observed in the c-Jun:c-Jun/AP-1 and c-Jun:c-Fos/AP-1 complexes (Kerppola and Curran, 1991a).
bHLH-ZIP proteins recognise the palindromic E-box DNA sequence motif (5' CANNTG
3') (Phillips, 1994). The crystal structure of the Max homodimer bound to its
cognate DNA sequence (Ferré-D'Amaré et al., 1993) reveals
that it forms a parallel, left-handed, four-helix bundle. The two basic-domain
helices project from the bundle and enter the major groove of the DNA binding
site from opposite directions in a fashion similar to that seen in GCN4
(Ellenberger et al., 1992; König and Richmond, 1993). The
-helices C-terminal to the basic
domain (termed H1) form the
four-helix bundle by packing against themselves and with the helices N-terminal
to the leucine zipper domains (termed H2). The H1 and H2 helices in each
monomer are linked via a structurally well defined eight residue loop. The
leucine zipper domain forms a regular coiled-coil structure.
Interestingly, the bHLH-ZIP proteins appear to bend DNA in a fashion similar to that seen for c-Jun and c-Fos (Kerppola and Curran, 1991a; 1991b; Fisher et al., 1992; Wechsler and Dang, 1992). It also appears that the bHLH-ZIP and bZIP families can associate with one another, as a bHLH-ZIP protein, termed FIP (Fos interacting protein), has been discovered which can heterodimerise with c-Fos in vitro (Blanar and Rutter, 1992). Cotransfection of FIP with c-fos into F9 cells produced transactivation of an AP-1 regulated reporter gene, however binding of the FIP:c-Fos complex to DNA has yet to be demonstrated in vitro (Blanar and Rutter, 1992).
In the case of bZIP homodimers, the dimer concentration, and thus the number of molecules bound to target DNA sequences, is dependent on both the monomer concentration and the protein dimerisation binding constant. If this is then extended to a mixture of bZIP monomers, the ratios of the various homo-/heterodimer species will be dependent on the relative concentrations of each of the monomers and all of the protein dimerisation binding constants for each of the different homo-/heterodimer combinations. A good example of this is the case of c-Jun and c-Fos. The dimerisation binding constant of c-Jun homodimers is only slightly less than that of c-Jun:c-Fos heterodimers, however, the dimerisation binding constant for c-Fos homodimers is very low. Thus, when c-Jun and c-Fos are mixed, c-Jun:c-Fos heterodimers are formed preferentially over c-Jun homodimers.
One further consequence of the varying affinities leucine zippers can have for one another is that they regulate the tightness of DNA binding (Alber, 1992). Thus, the lower the binding constant for a given leucine zipper pair, the lower will be the associated binding constant for the target DNA sequence. Therefore, in the mixed environment of the nucleus, altering the concentration of a single bZIP protein can not only alter the range of target sequences bound, but also how tightly each of them are bound by bZIP transcription factors. Thus, bZIP proteins are able to provide a very fine level of control over gene transcription.
Our current structural picture of how bZIP proteins interact with each other and DNA has largely been limited to what can be observed with X-ray diffraction techniques. These methods, although extremely useful, can only yield information on the crystalline form of protein and/or DNA. Crystalline forms of globular proteins usually adopt conformations that are very similar, if not identical, to the solution conformation. This can be rationalised by the large amount of solvent water trapped in protein crystals and the comparatively small number of contacts made between globular protein molecules in the crystal by comparison with the number of intramolecular contacts that hold the protein structure together.
However, bZIP domains are not globular and show a much larger number of crystallographic contacts which have an unknown effect on the structures observed. These contacts may have significant effects given the apparent flexibility of the components of bZIP/DNA complexes (e.g., DNA bending in the GCN4-bZIP:ATF/CREB complex; König and Richmond, 1993). X-ray diffraction is also, by its very nature, a static technique and has only a very limited ability to probe the dynamics of molecules. Thus it would be highly desirable to see the X-ray studies of GCN4-bZIP complemented by studies using a technique, such as NMR, which is able to yield both atomic resolution structural information as well as information on the dynamics of the molecule in its native solution state.
Currently, NMR studies on peptides encompassing elements of bZIP domains have provided only limited structural information (Oas et al., 1990; Saudek et al., 1990; 1991b; Junius et al., 1993). The key observation of these studies is that leucine zipper homodimers form symmetric structures in solution. Ironically, it is this same useful observation which has hampered further efforts to obtain more detailed structural information from these proteins using NMR. The symmetry seen in the NMR spectra of leucine zipper homodimers results in a problematic ambiguity which precludes the use of conventional structure calculation techniques. Thus, the current models of leucine zippers derived from NMR data are only of the monomer components of the dimers and hence are of almost no use in ascertaining the interactions which mediate dimerisation.
Therefore another key goal of this work has been to develop and apply new techniques which can yield complete dimer structures from the inherently ambiguous spectra of symmetric homodimers. The dimer structure of JunLZ thus obtained serves as the starting point for the realisation of further objectives. First the JunLZ structure represents another direct test of the Landschulz leucine zipper hypothesis (1988). Analysis of the structure in combination with biochemical data such as that obtained from mutational studies (e.g., O'Shea et al., 1992) can reveal both the molecular interactions responsible for the stability of the JunLZ dimer as well as those interactions responsible for the specificity of the interaction. Knowledge of these interactions provides a further insight into the regulation of eukaryotic gene expression. Furthermore, this knowledge can provide a basis for the rational design of both diagnostic kits and anticancer therapies based upon activity of the leucine zipper domain.